최근 몇 년간 전 세계를 강타했던 COVID-19 팬데믹 상황을 기억하실 겁니다. 매일같이 발표되는 확진자 수 뒤에는 감염 여부를 판정하는 진단 검사의 노력이 숨어있었는데요, 그 중심에 바로 역전사 중합효소연쇄반응(Reverse Transcription Polymerase Chain Reaction, RT-PCR)이라는 기술이 있었습니다. 이 기술 덕분에 우리는 눈에 보이지 않는 작은 바이러스의 존재를 빠르고 정확하게 확인할 수 있었지요.
그런데 여기서 한 가지 궁금증이 생깁니다. PCR은 원래 DNA를 증폭하는 기술로 알려져 있는데, SARS-CoV-2와 같은 RNA 바이러스는 어떻게 PCR로 검출할 수 있었을까요? 마치 영어로 쓰인 책의 내용을 복사하고 싶은데, 복사기가 한글만 인식하는 상황과 비슷하다고 할 수 있습니다. 이 문제를 해결하는 열쇠가 바로 '역전사(Reverse Transcription)' 과정에 숨어 있습니다.

이번 시간에는 우리 몸속 세포부터 바이러스까지, 생명 현상의 중요한 정보를 담고 있는 RNA를 분석하는 강력한 도구, RT-PCR의 기본 원리는 무엇인지, 실제 실험은 어떻게 진행되는지, 그리고 의학, 생명 과학 등 다양한 분야에서 어떻게 활용되는지 아주 깊이 있고 상세하게 파헤쳐 보겠습니다.
역전사 RNA 정보를 DNA로 번역
RT-PCR 기술의 가장 근본적인 원리는 바로 RNA를 주형(template)으로 사용하여 그 정보에 상보적인 DNA 가닥, 즉 cDNA(complementary DNA)를 만드는 '역전사' 과정과, 이렇게 만들어진 cDNA를 일반적인 PCR 기술로 대량 증폭하는 두 가지 핵심 단계를 순차적으로 결합한 것입니다. 왜 이런 복잡해 보이는 과정을 거쳐야 할까요? 그 이유는 PCR 기술의 핵심 엔진 역할을 하는 DNA 중합효소(DNA polymerase)라는 효소의 특성 때문입니다.
"잠깐만요, PCR은 DNA를 복사하는 기술이라고 들었는데, RNA는 왜 바로 복사하지 못하는 건가요? 그냥 RNA를 직접 증폭하는 효소를 쓰면 안 되나요?"
아주 예리한 질문입니다! 실제로 PCR에 사용되는 DNA 중합효소는 이름에서 알 수 있듯이 DNA 가닥을 주형으로 삼아서만 새로운 DNA 가닥을 합성할 수 있습니다. 마치 특정 언어(DNA)로 쓰인 문서만 복사할 수 있는 복사기와 같다고 할 수 있지요. RNA라는 다른 언어로 쓰인 문서는 직접 복사하지 못하는 것입니다. 따라서 우리가 RNA에 담긴 유전 정보를 PCR이라는 강력한 증폭 기술로 분석하려면, 먼저 RNA 정보를 DNA 중합효소가 인식할 수 있는 DNA 언어로 '번역'해주는 과정이 반드시 필요합니다.
이 중요한 번역가의 역할을 하는 것이 바로 역전사 효소(Reverse Transcriptase)입니다. 이 효소는 RNA 가닥을 주형으로 삼아 그에 상보적인 염기서열을 가진 DNA 가닥을 합성하는 놀라운 능력을 가지고 있습니다. 즉, RNA 정보를 DNA 정보로 변환시켜주는 것이지요. 이렇게 RNA로부터 만들어진 DNA를 특별히 상보적 DNA(complementary DNA), 줄여서 cDNA라고 부릅니다 [1]. 일단 cDNA가 만들어지면, 이제 이 cDNA는 일반적인 PCR의 주형으로 사용될 수 있습니다. 즉, DNA 중합효소가 이 cDNA를 인식하고 수많은 복사본을 만들어낼 수 있게 되는 것입니다.
결론적으로 RT-PCR은 RNA를 cDNA로 변환하는 역전사 단계와 cDNA를 증폭하는 PCR 단계라는 두 개의 핵심 모듈이 결합된 기술이라고 할 수 있습니다. 이 두 단계를 통해 우리는 불안정하고 직접 다루기 어려운 RNA 정보를 안정적인 DNA 형태로 변환하고, 이를 민감하게 검출하고 분석할 수 있는 강력한 수단을 얻게 되는 것입니다.
레트로바이러스에서 얻은 열쇠
그렇다면 RNA를 DNA로 바꾸는 이 놀라운 역전사 효소는 어디서 왔을까요? 놀랍게도 이 효소는 레트로바이러스(Retrovirus)라는 특별한 종류의 바이러스에서 처음 발견되었습니다. 레트로바이러스는 인간면역결핍 바이러스(HIV)처럼 자신의 유전 정보를 DNA가 아닌 RNA 형태로 가지고 있습니다. 이 바이러스가 숙주 세포에 침투하면, 자신이 가진 역전사 효소를 이용하여 RNA 유전 정보를 DNA 형태로 변환시킨 후, 이 DNA를 숙주 세포의 염색체 DNA에 슬쩍 끼워 넣습니다. 마치 스파이가 암호문을 해독하여 중요한 문서에 몰래 삽입하는 것과 비슷하다고 할 수 있지요.
과학자들은 레트로바이러스의 이러한 독특한 생존 전략에서 아이디어를 얻었습니다. 바이러스가 사용하는 역전사 효소를 분리하고 정제하여 실험실에서 RNA로부터 cDNA를 합성하는 데 성공한 것입니다 [14]. 이것이 바로 RT-PCR 기술 개발의 결정적인 돌파구가 되었습니다.
역전사 반응이 일어나기 위해서는 몇 가지 필수 구성 요소가 필요합니다. 먼저 당연히 주형이 될 RNA가 있어야 하고, 번역가 역할을 할 역전사 효소가 필요합니다. 그리고 역전사 효소가 DNA 합성을 시작할 지점을 알려주는 짧은 DNA 조각인 프라이머(primer), DNA의 기본 구성 단위인 dNTP(deoxynucleotide triphosphate: dATP, dGTP, dCTP, dTTP), 그리고 효소가 최적의 활성을 나타낼 수 있도록 환경을 조성해주는 완충액(buffer)과 마그네슘 이온(Mg²⁺) 등이 필요합니다.
프라이머 RNA 표적 설정
역전사 반응에서 프라이머는 어떤 종류를 사용하느냐에 따라 합성되는 cDNA의 종류와 특성이 달라지기 때문에 매우 중요합니다. 마치 어떤 종류의 지도를 사용하느냐에 따라 탐험의 범위와 목표가 달라지는 것과 같습니다. 주로 사용되는 프라이머에는 세 가지 종류가 있습니다.
첫 번째는 Oligo(dT) 프라이머입니다. 이 프라이머는 여러 개의 티민(T) 염기가 반복되는 구조(예: TTTTTTTTTTTTTTTTTT)를 가집니다. 진핵생물 mRNA의 특징 중 하나는 3' 말단에 아데닌(A) 염기가 길게 반복되는 폴리-A 꼬리(poly-A tail)를 가지고 있다는 점인데, Oligo(dT) 프라이머는 바로 이 폴리-A 꼬리에 상보적으로 결합하여 작동합니다. 따라서 이 프라이머를 활용하면 주로 성숙한 mRNA로부터 완전한 길이의 cDNA를 합성할 수 있어, 특정 단백질을 만드는 mRNA 정보 분석과 같은 유전자 발현 연구에 매우 유용합니다 [15]. 하지만 폴리-A 꼬리가 없는 RNA(예: 세균 mRNA, rRNA, tRNA, 일부 바이러스 RNA)나 분해되어 폴리-A 꼬리가 짧아진 mRNA에서는 cDNA 합성이 어렵다는 한계가 있습니다.
두 번째 종류는 Random 프라이머(Random hexamers 또는 nonamers)입니다. 이름에서 알 수 있듯이, 이 프라이머는 특정 서열 없이 무작위적인 염기서열로 구성된 짧은 DNA 조각(대개 6개 또는 9개 염기 길이)의 혼합물입니다. 이 프라이머들은 RNA 분자상의 여러 위치에 무작위적으로 결합하여 cDNA 합성을 개시합니다. 결과적으로 Oligo(dT) 프라이머와는 다르게 모든 종류의 RNA(mRNA, rRNA, tRNA, 바이러스 RNA 등)로부터 cDNA를 합성할 수 있습니다.
또한 RNA의 2차 구조가 복잡하거나 일부 분해된 RNA에서도 비교적 효율적으로 cDNA를 생성할 수 있다는 장점을 지닙니다 [16]. 이는 전체 RNA 집단을 대상으로 분석하거나 특정 RNA의 5' 말단 부위 정보가 중요할 때 유용하게 활용됩니다. 다만, 주로 짧은 길이의 cDNA 조각들이 생성되는 경향이 있습니다.
세 번째는 유전자 특이 프라이머(Gene-specific primer, GSP)입니다. 이 프라이머는 연구자가 분석하고자 하는 특정 유전자의 RNA 서열 중 일부와 정확히 상보적으로 결합하도록 특별히 설계됩니다. 그러므로 이 프라이머를 사용하면 오직 목표하는 특정 RNA로부터만 cDNA를 합성할 수 있다는 특징이 있습니다. 이는 반응의 특이도를 극도로 높여주므로, 특정 유전자의 존재 유무를 매우 민감하게 검출해야 하거나 전체 RNA 중에서 극미량으로 존재하는 특정 RNA를 분석하고자 할 때 매우 효과적입니다 [17]. 예를 들어, 특정 바이러스 유전자를 검출하는 진단 검사에서는 해당 바이러스 유전자에 특이적인 GSP가 사용됩니다. 그러나 다른 유전자의 RNA 정보는 얻을 수 없다는 제한점이 있습니다.
어떤 프라이머를 선택할지는 실험의 목적과 분석하려는 RNA의 종류에 따라 신중하게 결정해야 합니다. 때로는 Oligo(dT)와 Random 프라이머를 혼합하여 사용함으로써 각각의 장점을 취하고 단점을 보완하기도 합니다.
역전사 반응의 진행 단계
역전사 반응은 일반적으로 다음과 같은 단계로 진행됩니다. 첫째, 프라이머 결합(Primer Annealing) 단계입니다. 준비된 RNA 주형과 선택된 프라이머를 혼합하고, 프라이머가 RNA 주형의 특정 위치(Oligo(dT)는 폴리-A 꼬리, Random 프라이머는 무작위 위치, GSP는 특정 유전자 서열)에 효과적으로 결합할 수 있도록 적절한 온도로 조절합니다. 이 과정에서 RNA의 복잡한 2차 구조를 풀어주기 위해 초기에는 잠시 높은 온도(예: 65-70°C)로 처리한 후, 프라이머 결합에 적합한 낮은 온도로 낮추는 과정을 포함하기도 합니다.
둘째, cDNA 합성(cDNA Synthesis / Extension) 단계입니다. 이 단계에서는 역전사 효소와 dNTP 혼합물을 첨가하고, 역전사 효소가 최적의 활성을 나타내는 온도(일반적으로 42°C에서 55°C 사이이며, 사용하는 효소 종류에 따라 상이함)로 반응 온도를 조절합니다. 역전사 효소는 프라이머의 3' 말단부터 시작하여 RNA 주형을 따라 이동하면서 상보적인 dNTP를 순서대로 연결하여 새로운 DNA 가닥, 즉 첫 번째 가닥 cDNA(first-strand cDNA)를 합성해 나갑니다. 이 합성 과정에 필요한 시간은 목표하는 cDNA의 길이와 사용된 효소의 종류에 따라 달라지지만, 통상적으로 30분에서 1시간 정도 소요됩니다.
셋째, RNA 분해(RNA Degradation) 단계는 선택적으로 수행될 수 있습니다. 첫 번째 가닥 cDNA 합성이 완료되면 RNA와 DNA가 결합된 하이브리드 상태가 됩니다. 다음 단계인 PCR에서는 DNA 주형이 필요하므로, 원래의 RNA 주형 가닥을 제거하는 것이 반응 효율을 높이는 데 도움이 될 수 있습니다. 이를 위해 RNase H라는 효소를 처리하여 RNA:DNA 하이브리드에서 RNA 가닥만을 특이적으로 분해할 수 있습니다. 하지만 이 단계는 모든 RT-PCR 프로토콜에서 필수적인 것은 아닙니다.
마지막으로, 역전사 효소 불활성화(Enzyme Inactivation) 단계입니다. 역전사 반응이 종료된 후, 반응 온도를 충분히 높여서(대개 85°C에서 95°C 사이에서 5분 정도 처리) 역전사 효소의 활성을 완전히 제거합니다. 이는 후속 PCR 반응 단계에서 역전사 효소가 원치 않는 영향을 미치는 것을 방지하기 위한 중요한 과정입니다.
이렇게 모든 역전사 반응 단계가 완료되면, 원래의 RNA 정보는 이제 안정적인 cDNA 형태로 변환되어 다음 단계인 PCR 증폭 과정을 기다리게 됩니다.
cDNA 정보 대량 증폭
역전사 과정을 통해 성공적으로 얻어진 cDNA는 이제 일반적인 PCR 반응의 주형으로 사용될 준비가 되었습니다. PCR(Polymerase Chain Reaction)은 특정 DNA 영역을 기하급수적으로 증폭시키는 혁명적인 분자생물학 기술입니다 [2]. PCR 과정은 크게 세 가지 단계가 반복적으로 순환하면서 진행되며, 이를 통해 표적 DNA 서열을 대량으로 복제합니다.
첫 번째 단계는 변성(Denaturation)입니다. 반응 온도를 약 95°C로 높여 cDNA 이중나선(만약 두 번째 가닥 합성이 일어났거나, gDNA 오염이 있다면) 또는 cDNA:RNA 하이브리드 구조를 완전히 분리시켜 각각 단일 가닥으로 만듭니다. 이 과정은 다음 단계에서 프라이머가 표적 서열에 접근하여 결합할 수 있도록 길을 열어주는 필수적인 준비 단계입니다.
두 번째 단계는 결합(Annealing)입니다. 온도를 약 50°C에서 65°C 사이로 낮춥니다. 이 온도 범위는 증폭하고자 하는 특정 cDNA 영역의 양 끝에 결합하도록 특별히 설계된 한 쌍의 프라이머(Forward primer와 Reverse primer)가 각각의 상보적인 단일 가닥 cDNA 서열에 특이적으로 달라붙기에 적합합니다. 이 결합 온도는 사용하는 프라이머의 염기서열, 길이, 그리고 GC 함량에 따라 달라지며, PCR 반응의 특이성을 결정하는 매우 중요한 요소이므로 신중하게 최적화해야 합니다.
세 번째 단계는 신장(Extension/Elongation)입니다. 온도를 다시 약 72°C로 올립니다. 이 온도는 PCR 반응의 핵심 동력인 내열성 DNA 중합효소(thermostable DNA polymerase, 예: Taq polymerase)가 가장 활발하게 작용하는 최적 온도입니다. DNA 중합효소는 프라이머가 결합한 지점의 3' 말단부터 시작하여 단일 가닥 cDNA를 주형으로 삼아, 주변에 있는 dNTP들을 순서대로 가져와 붙여나가면서 새로운 상보적인 DNA 가닥을 합성합니다.
이 변성-결합-신장의 세 단계를 하나의 사이클(cycle)로 구성하여, 이 사이클을 일반적으로 25회에서 40회 정도 반복합니다. 각 사이클마다 표적 DNA 영역이 이론적으로 2배씩 증가하므로, 예를 들어 30 사이클을 반복하면 표적 DNA 분자가 $2^{30}$배, 즉 약 10억 배 이상으로 기하급수적으로 증폭됩니다. 이는 마치 작은 눈덩이를 언덕 위에서 굴리면 거대한 눈덩이가 되는 것처럼, 초기에는 미미했던 양의 cDNA 정보가 PCR 과정을 통해 검출 가능한 수준을 넘어 엄청난 양으로 늘어나는 원리입니다.
이처럼 RT-PCR은 RNA를 cDNA로 변환하는 역전사 과정과 cDNA를 기하급수적으로 증폭하는 PCR 과정의 절묘한 조합을 통해, 우리가 눈으로 볼 수 없는 미세한 RNA의 세계를 들여다볼 수 있게 해주는 강력한 현미경과 같은 역할을 수행합니다.
RT-PCR 과정
성공적인 RT-PCR 결과를 얻기 위해서는 단순히 원리를 이해하는 것을 넘어, 실험의 각 단계를 꼼꼼하게 수행하고 발생 가능한 문제점들을 미리 예방하는 것이 무엇보다 중요합니다. 특히 RNA는 DNA에 비해 매우 불안정하여 쉽게 분해될 수 있기 때문에, 실험 전 과정에 걸쳐 세심한 주의가 필요합니다. 마치 섬세한 유리 작품을 다루듯 조심스럽게 접근해야 하는 것이지요.
RT-PCR 실험 과정은 크게 RNA 추출 및 정제, cDNA 합성 (역전사), PCR 증폭, 그리고 결과 확인 및 분석의 네 단계로 나눌 수 있습니다. 각 단계별 핵심 내용과 주의사항을 자세히 살펴보겠습니다.
1단계 고품질 RNA 확보
모든 RT-PCR 실험의 성패는 양질의 RNA를 얼마나 잘 분리하고 정제하느냐에 달려있다고 해도 과언이 아닙니다. 아무리 좋은 시약과 장비를 사용하더라도 시작 물질인 RNA의 품질이 좋지 않다면 정확하고 신뢰성 있는 결과를 기대하기 어렵습니다. RNA 품질 저하의 가장 큰 주범은 바로 RNA 분해 효소(RNase)입니다.
"RNase가 그렇게 문제가 되나요? 그냥 조심하면 되는 거 아닌가요?"
그렇게 간단한 문제가 아닙니다. RNase는 우리 몸의 피부, 침, 땀은 물론이고 실험실 환경의 먼지, 공기 중 미생물 등 어디에나 존재합니다 [3]. 게다가 이 효소는 매우 안정적이어서 끓이거나(autoclaving) 일반적인 소독 방법으로도 쉽게 불활성화되지 않습니다. 따라서 실험자는 자신이 RNase 오염의 주된 원인이 될 수 있다는 사실을 항상 명심하고, RNA를 다루는 전 과정에서 RNase 오염을 막기 위한 철저한 노력을 기울여야 합니다.
RNase 오염을 최소화하기 위해서는 여러 핵심 수칙을 준수해야 합니다. 먼저, 실험에 사용하는 모든 소모품, 예를 들어 플라스틱 튜브, 피펫 팁, 시약 등은 반드시 'RNase-free' 인증을 받은 제품을 사용해야 합니다. 유리 기구의 경우, 사용 전 고온 처리(baking)나 DEPC(diethylpyrocarbonate) 용액 처리를 통해 RNase를 효과적으로 제거해야 합니다. 또한, 작업 공간의 청결 유지가 필수적입니다. 실험대는 사용 전후 RNase 제거 전용 스프레이(예: RNaseZap™)로 깨끗하게 닦아 오염원을 제거해야 합니다.
개인 보호 장비 착용도 중요합니다. 실험자는 항상 깨끗한 일회용 장갑을 착용하고, 실험 중에는 장갑 낀 손으로 얼굴, 머리카락, 휴대폰 등 잠재적 오염원을 만지지 않도록 각별히 주의해야 합니다. 마스크 착용 역시 침방울 등으로 인한 오염 가능성을 줄이는 데 도움이 될 수 있습니다. 실험 중에는 불필요한 대화나 움직임을 최소화하여 침이나 피부 세포가 날려 시료를 오염시키는 것을 방지해야 합니다.
가능하다면 RNA 작업만을 위한 전용 실험 공간(bench), 피펫 세트, 원심분리기 등을 확보하여 사용하는 것이 교차 오염을 막는 이상적인 방법입니다. 마지막으로, RNA 실험용 시약은 다른 실험과 분리하여 보관하고, 시약을 덜어낼 때는 반드시 새로운 RNase-free 팁을 사용하며, 사용하지 않을 때는 즉시 뚜껑을 닫아 공기 중 오염 가능성을 차단해야 합니다.
RNA 추출 방법은 실험 대상(세포, 조직, 혈액 등)과 연구 목적에 따라 다양한 기법이 활용됩니다. 가장 고전적이고 널리 알려진 방법은 Guanidinium thiocyanate-phenol-chloroform 추출법 (예: Trizol 시약 사용)이지만, 최근에는 사용이 간편하고 안전성이 높은 컬럼(column) 기반의 RNA 추출 키트가 보편적으로 사용되고 있습니다 [18]. 어떤 방법을 선택하든, 추출 과정 전반에 걸쳐 RNase 오염을 철저히 방지하고 RNA 분해를 최소화하는 것이 성공적인 RNA 추출의 핵심입니다. 추출된 RNA는 가능한 한 빨리 다음 실험에 사용하거나, 사용 전까지는 -70°C 이하의 초저온 냉동고에 에탄올 침전 상태 또는 RNase-free 물이나 완충액에 녹여 보관해야 합니다.
추출된 RNA는 다음 단계 실험 조건을 설정하고 결과의 신뢰성을 확보하기 위해 반드시 품질과 농도를 확인해야 합니다. RNA 농도 및 순도 측정에는 분광광도계(spectrophotometer)가 주로 사용됩니다. 260 nm 파장에서의 흡광도(A260)를 측정하여 RNA 농도를 계산하며 ($\text{RNA 농도 (µg/mL)} = A_{260} \times \text{희석 배수} \times 40$), 동시에 280 nm (단백질 흡수 파장)와 230 nm (유기용매, 염 등 기타 오염물질 흡수 파장)에서의 흡광도도 측정합니다.
이 값들을 이용하여 A260/A280 비율과 A260/A230 비율을 계산하는데, 이는 RNA의 순도를 평가하는 중요한 지표입니다. 순수한 RNA의 A260/A280 비율은 약 1.8 ~ 2.1 범위이며, 이 값보다 현저히 낮으면 단백질 오염을, 너무 높으면 페놀과 같은 다른 오염 물질의 존재를 의심할 수 있습니다. A260/A230 비율은 일반적으로 2.0 이상이어야 좋은 순도를 의미하며, 이 값보다 낮을 경우 추출 과정에서 사용된 유기 용매나 염(salt) 등이 잔류하여 오염되었을 가능성을 시사합니다 [4].
RNA의 농도 및 순도 확인과 더불어, RNA가 분해되지 않고 온전한 상태인지 확인하는 것도 매우 중요합니다. 이는 아가로스 겔 전기영동(agarose gel electrophoresis)을 통해 시각적으로 평가할 수 있습니다. 포름알데히드 변성 겔(formaldehyde denaturing gel)을 사용하거나, Agilent Bioanalyzer와 같은 상용화된 RNA 분석 시스템을 이용하면 더욱 정확한 평가가 가능합니다.
총 RNA(total RNA)를 전기영동했을 때, 진핵생물의 경우 크기가 큰 28S rRNA와 작은 18S rRNA 밴드가 뚜렷하게 나타나야 하며, 이 두 밴드의 비율이 약 2:1 정도일 때 온전한 RNA로 간주합니다. 만약 이 밴드들이 희미하거나 전체적으로 끌리는 양상(smear)을 보인다면 RNA가 분해되었음을 의미하며, 이러한 RNA는 실험 결과의 정확도를 심각하게 저해할 수 있습니다. 최근에는 RNA 품질을 객관적인 수치로 평가하는 RNA Integrity Number (RIN)이라는 지표가 널리 사용되며, 일반적으로 RIN 값이 7 이상이면 좋은 품질의 RNA로 간주합니다 [19].
2단계 cDNA 합성
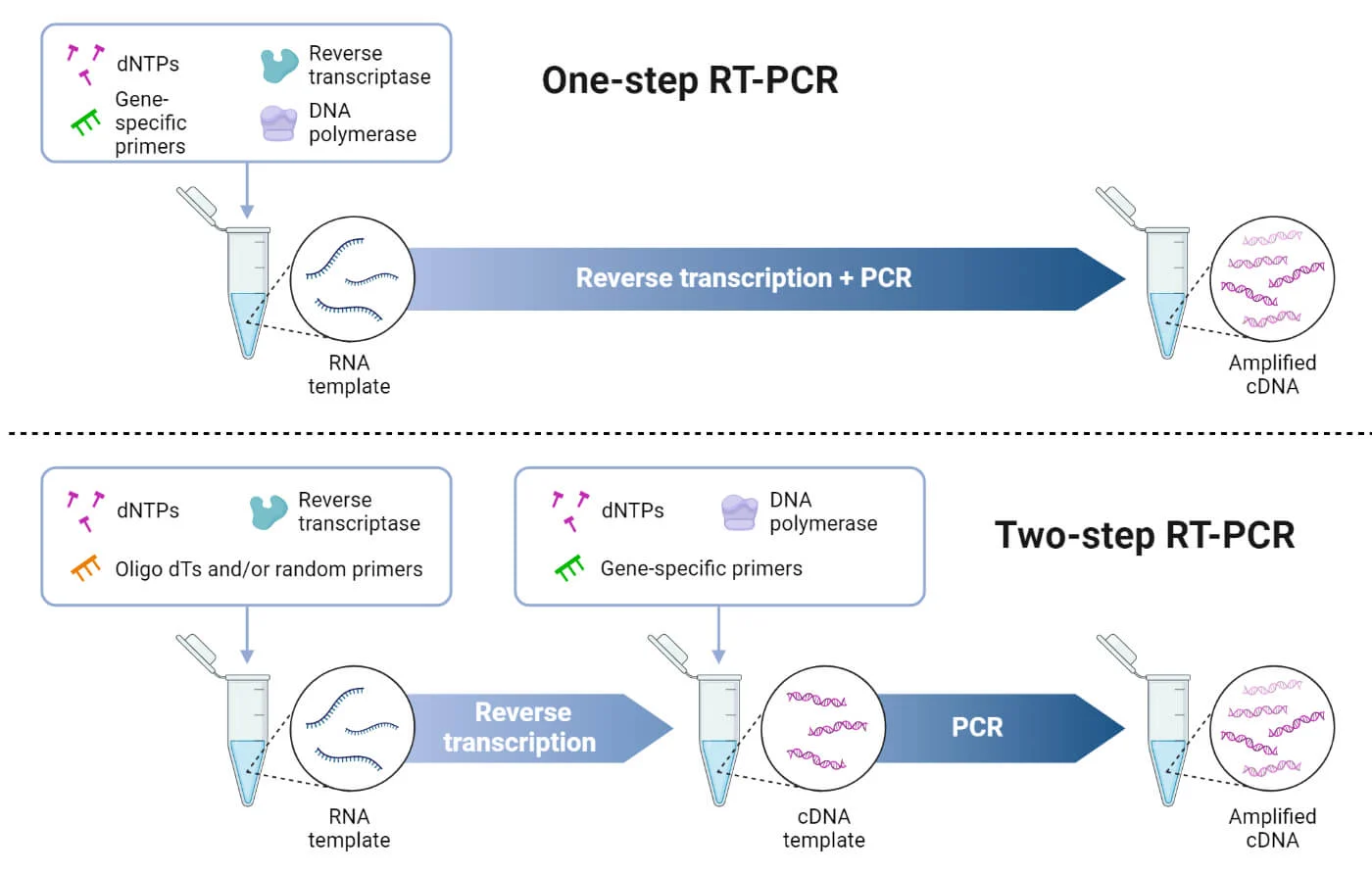
고품질의 RNA가 성공적으로 준비되었다면, 이제 역전사 반응을 통해 cDNA를 합성할 차례입니다. 앞서 원리 부분에서 설명했듯이, 이 단계에서는 RNA 주형, 역전사 효소, 적절한 프라이머, dNTP, 그리고 반응 완충액 등이 필요합니다. 이 과정에서 중요한 결정 사항 중 하나는 One-step RT-PCR을 수행할지, 아니면 Two-step RT-PCR을 수행할지를 선택하는 것입니다.
Two-step RT-PCR 방식은 이름에서 알 수 있듯이 역전사 반응과 PCR 반응을 별도의 튜브에서 순차적으로 진행합니다. 먼저 RNA로부터 cDNA를 합성하는 역전사 반응을 수행하고 (첫 번째 단계), 이렇게 생성된 cDNA 용액의 일부를 조심스럽게 덜어내어 새로운 튜브에서 PCR 반응을 진행합니다 (두 번째 단계). 이 방식은 몇 가지 장점을 가집니다.
첫째, 유연성이 높습니다. 합성된 cDNA는 안정적으로 보관이 가능하며, 필요에 따라 여러 번의 다른 PCR 반응(예: 다른 유전자 분석, 다른 PCR 조건 테스트)에 나누어 사용할 수 있습니다. 이는 마치 원본 문서를 복사해 두고 필요할 때마다 꺼내 쓰는 것과 유사합니다.
둘째, 최적화가 용이합니다. 역전사 단계와 PCR 단계를 분리하여 진행하므로 각각의 반응 조건을 독립적으로 최적화하기 수월합니다. 예를 들어, 역전사 단계에서는 Random 프라이머를 사용하고 PCR 단계에서는 유전자 특이 프라이머를 사용하는 등 다양한 조합을 자유롭게 시도할 수 있습니다.
셋째, 다중 분석에 유리합니다. 하나의 cDNA 샘플로부터 여러 종류의 유전자 발현을 동시에 분석하거나, 서로 다른 PCR 조건을 적용하여 비교 분석하는 데 적합합니다. 반면, 단점으로는 두 단계를 거치므로 One-step 방식보다 시간이 더 오래 걸리고, 튜브 간 시료 이동 과정에서 외부 오염(contamination)이나 피펫팅 오류로 인한 시료 손실(pipetting error)의 위험이 상대적으로 높으며, 두 단계 반응을 위한 별도의 시약과 튜브 등이 필요하여 비용이 더 들 수 있습니다. 따라서 Two-step RT-PCR은 여러 유전자의 발현을 동시에 비교 분석하는 연구, 합성된 cDNA를 장기간 보관하며 반복적으로 사용해야 하는 경우, 각 반응 단계의 세밀한 최적화가 필요한 경우 등에 주로 선택됩니다.
반면, One-step RT-PCR 방식은 역전사 반응과 PCR 반응을 하나의 튜브 안에서 연속적으로 진행합니다. 반응 튜브에는 역전사 효소와 DNA 중합효소가 모두 포함된 특수 혼합 시약(master mix)과 RNA 주형, 프라이머, dNTP 등을 함께 넣고, 온도 사이클 프로그램을 설정하여 역전사 반응과 후속 PCR 반응이 순차적으로 일어나도록 합니다. 이 방식의 가장 큰 장점은 간편함과 신속성입니다. 모든 반응이 한 튜브에서 이루어지므로 실험 과정이 매우 단순하고 빠르게 완료될 수 있습니다.
또한, 튜브를 열고 닫거나 시료를 옮기는 횟수가 최소화되므로 외부 오염이나 교차 오염(cross-contamination)의 위험이 현저히 낮습니다. 이는 특히 많은 수의 시료를 신속하게 처리해야 하는 진단 검사 환경에서 매우 중요한 이점입니다. 더불어 실험 과정이 단순하여 자동화 장비를 이용한 고처리량(High-throughput) 분석에도 유리합니다. 하지만 단점도 존재합니다. 역전사 반응과 PCR 반응이 동일한 완충액 조건 하에서 진행되기 때문에, 각 단계에 대한 개별적인 최적화가 어렵습니다.
이는 두 효소가 최적으로 작동하는 조건이 서로 다를 수 있기 때문입니다. 또한, 합성된 cDNA를 다른 실험에 재사용하기 어렵고, 주로 유전자 특이 프라이머(GSP)를 사용해야 하므로 한 번의 반응으로 하나의 표적 유전자만 분석할 수 있어 유연성이 낮습니다. 경우에 따라서는 Two-step 방식보다 민감도가 다소 낮을 수 있다는 점도 고려해야 합니다. One-step RT-PCR은 특정 유전자의 존재 유무를 빠르게 확인하는 스크리닝, 대량의 임상 시료를 신속하게 분석해야 하는 진단 검사(예: COVID-19 진단), 그리고 오염에 매우 민감한 실험 등에 널리 사용됩니다.
어떤 방식을 선택하든, 반응 조건의 최적화는 성공적인 결과를 위해 필수적입니다. 특히 역전사 온도는 RNA의 2차 구조를 풀어주고 프라이머 결합 효율 및 효소 활성에 직접적인 영향을 미치므로 중요하게 고려해야 합니다. 사용하는 역전사 효소의 종류(예: M-MLV, AMV 유래 효소 또는 유전공학적으로 개량된 고온 활성 효소)에 따라 최적 온도가 다르므로, 제조사의 권장 사항을 따르는 것이 기본입니다. 이 외에도 RNA 주형의 양, 프라이머의 종류와 농도, 반응 시간 등 여러 요인이 결과에 영향을 미치므로, 필요하다면 예비 실험을 통해 최적 조건을 찾는 과정이 요구될 수 있습니다.
3단계 PCR 증폭 표적 cDNA 복제
역전사 반응(Two-step 방식의 경우) 또는 One-step 반응의 PCR 단계에서는 앞서 생성된 cDNA를 주형으로 삼아, 연구자가 원하는 특정 영역을 대량으로 증폭시킵니다. 성공적인 PCR 증폭을 위해서는 몇 가지 핵심 요소를 신중하게 고려하고 최적화하는 과정이 필요합니다.
PCR 반응을 위해서는 필수적인 구성 요소들이 필요합니다. 여기에는 주형이 되는 cDNA, 증폭하고자 하는 영역의 양쪽 끝 서열에 특이적으로 결합하는 한 쌍의 프라이머(Forward & Reverse), 고온에서도 안정적인 내열성 DNA 중합효소(DNA polymerase), DNA의 기본 구성 단위인 dNTP 혼합물, 효소 활성에 필수적인 MgCl₂, 그리고 적절한 pH와 염 농도를 유지하여 반응 환경을 조성하는 완충액(buffer)이 포함됩니다. 최근에는 이러한 구성 요소들이 미리 최적의 비율로 혼합된 PCR 마스터 믹스(master mix) 형태의 제품이 널리 사용되어, 실험의 편의성을 높이고 각 반응 간의 편차를 줄여 재현성을 향상시키는 데 기여하고 있습니다.
프라이머 설계는 PCR 결과의 특이성(specificity)과 효율성(efficiency)을 결정짓는 가장 중요한 요소 중 하나입니다. 잘 설계된 프라이머는 목표하는 cDNA 서열에만 정확하게 결합하고 다른 유사 서열에는 결합하지 않아(높은 특이성), 원치 않는 부산물 없이 목표 산물만을 효율적으로 증폭시켜야 합니다(높은 효율성). 좋은 프라이머를 설계하기 위한 일반적인 가이드라인은 여러 측면을 고려합니다.
길이(Length)는 보통 18~25 염기(base pair, bp) 정도가 적절하며, 너무 짧으면 특이성이 떨어지고 너무 길면 결합 효율이 낮아질 수 있습니다. 녹는점(Melting Temperature, Tm)은 프라이머가 주형 DNA의 절반과 결합하는 온도를 의미하며, PCR의 결합 온도(annealing temperature) 설정에 중요한 기준이 됩니다. Forward와 Reverse 프라이머의 Tm 값은 서로 유사해야 하고(보통 5°C 이내 차이), 일반적으로 55°C ~ 65°C 범위에 있도록 설계합니다. Tm 값은 프라이머의 염기 구성(GC 함량)과 길이에 따라 달라지며, 다양한 계산 공식이나 소프트웨어를 이용해 예측할 수 있습니다.
GC 함량(GC Content), 즉 프라이머 서열 내 구아닌(G)과 시토신(C) 염기의 비율은 보통 40~60% 범위가 적절하며, 프라이머의 안정성과 Tm 값에 영향을 줍니다. 설계된 프라이머의 서열 특이성(Specificity)은 BLAST와 같은 서열 유사성 검색 도구를 사용하여 목표 유전자 외에 다른 유전자나 반복 서열에 결합할 가능성이 없는지 반드시 검증해야 합니다. 마지막으로, 프라이머 자체적으로 헤어핀(hairpin)이나 루프(loop) 같은 내부 2차 구조를 형성하거나, 프라이머끼리 서로 결합하는 자가 이합체(self-dimer) 또는 Forward와 Reverse 프라이머가 서로 결합하는 교차 이합체(cross-dimer)를 형성할 가능성을 최소화해야 합니다. 이러한 구조는 프라이머가 표적 DNA에 결합하는 것을 방해하여 PCR 효율을 저해할 수 있습니다. Primer3, OligoAnalyzer와 같은 다양한 프라이머 설계 전용 소프트웨어를 활용하면 이러한 요소들을 종합적으로 평가하여 최적의 프라이머를 설계하는 데 큰 도움이 됩니다 [5, 20].
최적의 프라이머가 준비되었다면, 이제 효율적이고 특이적인 증폭을 얻기 위해 PCR 반응 조건을 최적화해야 합니다. 주요 최적화 대상은 결합 온도(Annealing Temperature, Ta)입니다. 이는 PCR 특이성에 가장 큰 영향을 미치는 요소로서, 너무 낮으면 프라이머가 표적 서열 외의 비특이적인 부위에도 결합하여 원치 않는 PCR 산물이 생성될 수 있고, 반대로 너무 높으면 프라이머가 표적 서열에 제대로 결합하지 못해 PCR 산물이 생성되지 않거나 효율이 크게 떨어질 수 있습니다.
최적의 Ta는 보통 프라이머 쌍의 계산된 Tm 값보다 약 3 ~
5°C 낮게 설정하지만, 경험적으로 여러 온도를 테스트하여(예: gradient PCR 사용) 가장 깨끗하고 강한 증폭이 일어나는 온도를 찾는 것이 가장 확실한 방법입니다. 사이클 수(Number of Cycles)는 증폭되는 PCR 산물의 양을 결정합니다. 너무 적으면 검출이 어렵고, 너무 많으면 반응이 포화 상태에 이르러 정량성이 떨어지거나 비특이적 산물이 축적될 수 있으므로, 보통 25~
40 사이클 범위에서 실험 목적에 맞게 설정합니다.
신장 시간(Extension Time)은 DNA 중합효소가 새로운 DNA 가닥을 합성하는 데 필요한 시간으로, 증폭하려는 PCR 산물의 길이와 사용하는 DNA 중합효소의 종류(합성 속도)에 따라 조절합니다. 일반적으로 Taq polymerase의 경우 1 kb당 1분 정도를 기준으로 합니다. 마지막으로, 효소 및 MgCl₂ 농도 역시 PCR 효율과 특이성에 영향을 미칠 수 있으므로, 필요에 따라 최적화 대상이 될 수 있습니다. 특히 MgCl₂ 농도는 프라이머 결합과 효소 활성에 중요하며, 농도가 너무 낮으면 효율이 저하되고 너무 높으면 비특이적 증폭이 증가할 수 있습니다.
4단계 결과 확인 및 분석
PCR 증폭이 완료되면, 마지막 단계는 원하는 표적 cDNA가 제대로 증폭되었는지, 그리고 그 양은 어느 정도인지를 확인하고 분석하는 것입니다. 분석 방법은 크게 두 가지 접근 방식으로 나눌 수 있습니다.
첫 번째는 가장 전통적이고 기본적인 방법인 겔 전기영동(Gel Electrophoresis)입니다. 이 방법은 증폭된 PCR 산물을 아가로스 겔(agarose gel)이라는 다공성 매트릭스에 주입하고 전류를 흘려보내는 원리를 이용합니다. 음전하(-)를 띤 DNA 분자들은 양극(+) 방향으로 이동하는데, 이때 DNA 분자의 크기(길이)에 따라 이동 속도가 달라집니다.
즉, 크기가 작을수록 겔의 그물 구조를 더 쉽게 통과하여 멀리 이동하고, 크기가 클수록 이동 속도가 느려집니다. 미리 크기를 알고 있는 DNA 크기 마커(DNA size marker)를 함께 전기영동하여 그 이동 거리를 비교하면, 증폭된 PCR 산물의 크기를 정확하게 확인할 수 있습니다. 또한, 겔을 EtBr(Ethidium Bromide)이나 SYBR Safe와 같은 DNA 염색 시약으로 염색한 후 UV 광선을 비추면 DNA 밴드가 형광을 내며 시각화되는데, 이 밴드의 위치를 통해 예상된 크기의 산물이 증폭되었는지 확인하고 밴드의 밝기(intensity)를 통해 증폭된 DNA의 양을 대략적으로 추정할 수 있습니다.
겔 전기영동의 장점은 비교적 간단하고 저렴한 장비로 결과를 확인할 수 있으며, 증폭 산물의 크기를 직접 확인할 수 있어 비특이적 증폭 여부(예상치 못한 크기의 밴드 생성)를 판단하는 데 유용하다는 점입니다. 그러나 정량 분석에는 한계가 있습니다. 밴드의 밝기는 증폭 양과 어느 정도 비례하지만, PCR 반응 후반부의 포화 효과(plateau effect) 때문에 초기 주형 양을 정확하게 반영하지 못할 수 있습니다. 또한, 감도가 상대적으로 낮아 미량의 증폭 산물을 확인하기 어려울 수 있으며, 분석 과정에 시간이 소요되고 EtBr과 같은 유해 시약을 사용해야 하는 경우도 있습니다. 주로 Endpoint RT-PCR (PCR 반응 종료 후 분석) 결과 확인에 사용됩니다.
두 번째 분석 방법은 실시간 PCR(Real-time PCR, qPCR)입니다. 이 기술은 PCR 증폭 과정 중에 생성되는 DNA 양을 실시간으로 모니터링하여 정량적으로 분석하는 혁신적인 방법입니다. 이를 위해 형광 물질을 이용하는데, 대표적으로 두 가지 방식이 널리 사용됩니다. 하나는 SYBR Green 방식입니다. SYBR Green I이라는 형광 염료는 DNA 이중나선(double-strand DNA)에 특이적으로 결합했을 때만 강한 형광을 방출합니다.
따라서 PCR 사이클이 진행됨에 따라 이중나선 DNA 산물이 기하급수적으로 증가하면서 형광 신호 역시 강해집니다. 이 방식은 비교적 저렴하고 어떤 유전자든 프라이머만 있으면 사용할 수 있다는 장점이 있지만, 비특이적 증폭 산물이나 프라이머 이합체(primer dimer)도 이중나선 구조를 가지므로 형광 신호를 발생시켜 위양성(false positive) 결과를 유발할 수 있다는 단점이 있습니다.
따라서 반응 종료 후 용융 곡선 분석(melting curve analysis)을 통해 증폭된 산물의 특이성을 확인하는 과정이 필수적으로 동반되어야 합니다. 다른 하나는 프로브(Probe) 방식 (예: TaqMan probe)입니다. 이 방식은 특정 유전자 서열 내부에 결합하는 형광 표지된 짧은 DNA 조각(probe)을 추가로 사용합니다. 프로브의 한쪽 끝에는 형광 물질(reporter dye)이, 다른 쪽 끝에는 이 형광을 억제하는 물질(quencher dye)이 붙어 있습니다.
평소에는 quencher가 reporter의 형광을 억제하고 있지만, PCR 신장 단계에서 DNA 중합효소가 프로브가 결합한 부위를 지나가면서 프로브를 분해하면(5'->3' exonuclease activity), reporter 형광 물질이 quencher로부터 떨어져 나와 비로소 형광 신호를 내게 됩니다. 이 방식은 표적 유전자 서열에만 특이적으로 결합하는 프로브를 사용하므로 SYBR Green 방식보다 특이성이 훨씬 높다는 장점이 있으나, 각 유전자마다 별도의 프로브를 설계하고 합성해야 하므로 비용이 더 많이 든다는 단점이 있습니다 [6].
역전사(RT) 과정과 실시간 PCR(qPCR)을 결합한 RT-qPCR (Quantitative Real-time RT-PCR)은 RNA의 초기 양을 매우 정확하고 민감하게 정량 분석할 수 있는 강력한 기술입니다. qPCR 장비는 각 PCR 사이클마다 발생하는 형광 신호를 실시간으로 측정하여 증폭 곡선(amplification plot)을 그립니다. 이 곡선에서 형광 신호가 미리 설정된 임계값(threshold)을 넘어서는 시점의 사이클 수를 Ct 값(Cycle threshold value) 또는 Cq 값(Quantification cycle value)이라고 정의합니다.
중요한 점은, Ct 값은 초기 주형 RNA(또는 그로부터 생성된 cDNA)의 양과 반비례한다는 것입니다. 즉, 초기 RNA 양이 많을수록 더 적은 사이클 만에 임계값에 도달하므로 Ct 값은 작아지고, 초기 RNA 양이 적을수록 더 많은 사이클이 필요하므로 Ct 값은 커집니다. 이 원리를 이용하여 미지의 시료 속 RNA 양을 이미 농도를 알고 있는 표준 RNA(standard RNA)의 Ct 값과 비교하여 절대 정량하거나, 서로 다른 샘플 간(예: 처리군 vs 대조군)의 RNA 양 차이를 상대적으로 비교하는 상대 정량 분석을 수행할 수 있습니다. 특히 유전자 발현 변화 연구에 가장 널리 사용되는 핵심적인 방법론입니다.
마지막으로, 증폭된 PCR 산물이 정말 우리가 목표했던 유전자의 서열과 일치하는지 최종적으로 확인하는 가장 확실한 방법은 염기서열 분석(Sequencing)입니다. 특히 새로운 유전자를 클로닝하거나, 돌연변이의 존재를 확인하거나, 겔 전기영동 등에서 비특이적 증폭이 의심될 때 염기서열 분석을 통해 정확한 유전 정보를 얻을 수 있습니다.
아래 표는 지금까지 설명한 주요 RT-PCR 방법들의 특징을 요약한 것입니다.
| 구분 | 특징 | 장점 | 단점 | 주요 용도 |
|---|---|---|---|---|
| Two-step RT-PCR | 역전사, PCR 분리 진행 | cDNA 재사용 가능, 각 단계 최적화 용이, 유연성 높음 | 시간 소요, 오염/손실 위험 증가 | 여러 유전자 분석, cDNA 라이브러리 제작, 유전자 발현 연구 |
| One-step RT-PCR | 역전사, PCR 한 튜브에서 연속 진행 | 간편, 신속, 오염 위험 감소, 대량 처리 용이 | 각 단계 최적화 어려움, cDNA 재사용 어려움, 유연성 낮음 | 특정 유전자 유무 확인, 고처리량 스크리닝, 진단 검사 |
| Endpoint RT-PCR | RT + 일반 PCR (겔 전기영동 분석) | 상대적으로 저렴한 장비/시약, 간단한 실험 과정, 크기 확인 용이 | 정량 분석 어려움, 민감도/특이도 상대적 낮음, 후처리 분석 필요 | 유전자 존재 유무 확인, 클로닝 위한 유전자 증폭, RNA splicing 분석 |
| RT-qPCR | RT + 실시간 PCR (형광 기반 정량 분석) | RNA 정량 분석 가능, 높은 민감도와 특이도, 넓은 검출 범위 | 고가 장비 필요, 실험 설계 및 데이터 분석 복잡성 증가, Ct 값 해석 주의 필요 | 유전자 발현 정량 분석, 바이러스 정량 검출, microRNA 분석 |
이처럼 RT-PCR 실험은 여러 단계로 구성되며, 각 단계마다 성공적인 결과를 얻기 위한 핵심 원리와 주의사항들이 있습니다. 다음으로는 이렇게 강력한 RT-PCR 기술이 실제 연구와 진단 현장에서 어떻게 활용되고 있는지 구체적인 사례들을 통해 알아보겠습니다.
RT-PCR의 주요 활용 분야
RT-PCR 기술은 RNA라는 생명의 언어를 해독하는 강력한 능력을 바탕으로, 현대 생명 과학 연구와 의학 진단 분야에서 없어서는 안 될 핵심적인 도구로 자리매김했습니다. 마치 만능 열쇠처럼, 유전자의 발현 조절 비밀을 풀고, 질병의 원인을 밝혀내며, 감염병을 신속하게 진단하는 등 다양한 문제 해결에 결정적인 역할을 수행하고 있습니다. 그 활약상을 구체적인 분야별로 살펴보겠습니다.
유전자 발현 분석
세포 내에서 특정 유전자가 얼마나 활발하게 작동하고 있는지를 알아보는 유전자 발현 분석은 생명 현상의 근본 원리를 이해하는 데 매우 중요합니다. 유전 정보는 DNA에 저장되어 있지만, 실제로 세포의 기능 수행에 필요한 단백질을 만들려면 먼저 DNA 정보가 mRNA(messenger RNA)로 전사(transcription)되는 과정을 거쳐야 합니다. 따라서 세포 내 특정 mRNA의 양을 측정하면, 해당 유전자가 얼마나 활발하게 단백질 생산을 위해 '일하고 있는지'를 간접적으로 알 수 있습니다.
바로 이 mRNA 양을 정확하게 측정하는 데 가장 널리 사용되는 기술이 바로 RT-qPCR입니다. 특정 조건(예: 약물 처리, 특정 성장 단계, 질병 상태 등)에 따라 관심 있는 유전자의 mRNA 발현량이 어떻게 변하는지를 대조군과 비교하여 정량적으로 분석할 수 있습니다. 예를 들어 신약 개발 연구에서 새로운 항암제의 효과를 평가할 때, 약물을 처리한 암세포와 처리하지 않은 암세포에서 세포 주기 조절 유전자(예: Cyclin D1, p21)나 세포 사멸 관련 유전자(예: Bax, Bcl-2)의 mRNA 발현량을 RT-qPCR로 측정하여 비교할 수 있습니다.
만약 약물 처리군에서 세포 성장 촉진 유전자인 Cyclin D1 mRNA는 감소하고, 세포 주기 억제 유전자인 p21*이나 세포 사멸 유도 유전자인 *Bax mRNA는 증가한다면, 이 약물이 이러한 유전자들의 발현 조절을 통해 항암 효과를 나타낼 수 있다는 강력한 증거가 됩니다 [7]. 또한, 배아 발생 과정이나 줄기세포가 특정 세포(예: 신경세포, 근육세포)로 분화하는 과정에서 어떤 유전자들이 시기별로 켜지고 꺼지는지를 추적하는 연구에도 RT-qPCR이 필수적으로 사용됩니다. 특정 시점에서 특정 전사 인자(transcription factor)나 세포 표지 유전자(marker gene)의 mRNA 발현량을 측정함으로써 발생 및 분화 과정을 조절하는 핵심 유전자 네트워크를 파악할 수 있습니다.
이처럼 RT-qPCR을 이용한 유전자 발현 분석은 생명 현상의 다양한 질문에 답을 제공하는 기본적인 연구 방법론으로 활용되고 있습니다.
감염성 질환 진단
RT-PCR 기술의 진가가 가장 극적으로 드러난 분야 중 하나는 바로 감염성 질환 진단, 특히 RNA 바이러스 감염 진단입니다. 앞서 언급했듯이, COVID-19 팬데믹 상황에서 SARS-CoV-2 바이러스 감염 여부를 신속하고 정확하게 진단하는 데 RT-qPCR 검사가 결정적인 역할을 했습니다 [8].
그 원리는 비교적 간단합니다. 환자의 검체(예: 코나 목에서 채취한 비인두 도말액, 침, 객담 등)에서 RNA를 추출한 뒤, 해당 바이러스만이 가지고 있는 특이적인 유전자 서열(예: SARS-CoV-2의 E 유전자, RdRp 유전자, N 유전자 등)을 표적으로 하는 프라이머와 프로브를 이용하여 One-step RT-qPCR을 수행합니다. 만약 검체 내에 바이러스 RNA가 존재한다면, RT-qPCR 반응을 통해 해당 유전자가 증폭되면서 형광 신호가 발생하고, Ct 값이 특정 기준치 이하로 낮게 측정됩니다. 이를 통해 감염 여부를 판정하는 것입니다.
RT-PCR 기반 진단법의 가장 큰 장점은 매우 높은 민감도입니다. 감염 초기처럼 바이러스의 양이 매우 적을 때에도 검출이 가능하며, 배양 검사처럼 시간이 오래 걸리지 않고 몇 시간 내에 결과를 얻을 수 있습니다. 이러한 신속성과 정확성 덕분에 RT-PCR은 호흡기 바이러스(인플루엔자, RSV, 코로나바이러스 등), 혈액 매개 바이러스(HIV, HCV, HBV 등), 기타 바이러스(노로바이러스, 로타바이러스, 지카 바이러스 등)를 포함한 다양한 RNA 바이러스 감염 진단에 표준 검사법으로 사용되고 있습니다. 뿐만 아니라 세균 감염 진단에도 활용될 수 있는데, 예를 들어 특정 환경 조건에서만 발현되는 독소 유전자의 mRNA를 RT-PCR로 검출하여 병원성 세균의 존재나 활성 상태를 판단하는 데 도움을 줄 수 있습니다.
유전 질환 진단 및 연구
유전 질환은 DNA 염기서열 자체의 돌연변이로 인해 발생하는 경우가 많지만, 때로는 DNA 서열은 정상이더라도 RNA 단계에서 문제가 발생하여 질병으로 이어지기도 합니다. 예를 들어, 비정상적인 RNA 스플라이싱(splicing)은 유전 질환의 중요한 원인 중 하나입니다. 스플라이싱은 유전자 정보가 mRNA로 전사된 후, 단백질을 코딩하지 않는 부위인 인트론(intron)은 제거되고 단백질 코딩 부위인 엑손(exon)들만 연결되는 과정입니다. 이 과정에 오류가 생겨 특정 엑손이 빠지거나(exon skipping), 인트론의 일부가 포함되거나(intron retention), 다른 엑손이 잘못 끼어들어가면(alternative splicing error), 결국 비정상적인 단백질이 만들어지거나 단백질이 아예 만들어지지 않아 질병을 유발할 수 있습니다 [9].
RT-PCR은 이러한 RNA 스플라이싱 오류를 검출하는 데 매우 유용합니다. 환자의 세포나 조직에서 RNA를 추출하여 관심 유전자의 mRNA를 cDNA로 변환한 뒤, 특정 엑손 주변 부위를 증폭하는 프라이머를 이용하여 PCR을 수행합니다. 만약 스플라이싱 오류로 인해 엑손이 빠졌다면 정상보다 짧은 길이의 PCR 산물이 증폭될 것이고, 인트론이 포함되었다면 더 긴 길이의 산물이 증폭될 것입니다.
이렇게 증폭된 PCR 산물의 크기를 겔 전기영동으로 비교하거나 염기서열 분석을 통해 스플라이싱 오류 여부와 그 패턴을 정확하게 확인할 수 있습니다. 예를 들어, 척수성 근위축증(SMA) 치료제의 효과를 평가하기 위해 환자 세포에서 SMN2 mRNA의 스플라이싱 패턴 변화를 RT-PCR로 분석하는 것이 중요합니다 [21]. 또한, 특정 유전 질환에서 관련 유전자의 mRNA 발현량이 비정상적으로 증가하거나 감소하는 경우도 있는데, 이는 RT-qPCR을 통해 정확하게 측정하여 진단 및 연구에 활용할 수 있습니다.
암 연구 및 진단
암은 유전자 변이와 비정상적인 유전자 발현 조절이 복합적으로 작용하여 발생하는 질병입니다. RT-PCR은 암의 발생, 진행, 전이 과정에 관여하는 다양한 유전자들의 발현 변화를 분석하고, 암 진단 및 예후 예측에 활용될 수 있는 RNA 바이오마커를 검출하는 데 핵심적인 역할을 합니다.
암 관련 유전자 발현 분석을 위해 암 조직과 주변 정상 조직에서 특정 암 유전자(oncogene)나 종양 억제 유전자(tumor suppressor gene)의 mRNA 발현량을 RT-qPCR로 비교 분석하여 암의 특징을 파악하고 치료 전략 수립에 활용합니다. 예를 들어, 유방암에서 HER2 유전자의 과발현 여부를 확인하는 것은 표적 치료제(허셉틴) 사용 여부를 결정하는 데 중요합니다 [10].
또한, 융합 유전자 검출에도 RT-PCR이 활용됩니다. 특정 암에서는 염색체 이상으로 두 개의 다른 유전자가 합쳐져 융합 유전자(fusion gene)가 만들어지는데, 만성 골수성 백혈병(CML)의 BCR-ABL 융합 유전자가 대표적인 예입니다. 환자의 혈액 세포에서 RNA를 추출하여 BCR-ABL 융합 부위를 표적으로 하는 RT-PCR(주로 RT-qPCR)을 수행하면, CML을 매우 민감하고 특이적으로 진단할 수 있을 뿐만 아니라, 치료 후 남아있는 미세 잔존 질환(MRD)을 정량적으로 모니터링하여 재발 위험 예측 및 치료 방향 결정에 결정적인 정보를 제공합니다 [11].
최근에는 액체 생검(Liquid Biopsy) 분야에서도 RT-PCR이 주목받고 있습니다. 혈액이나 소변과 같은 체액 속에 떠다니는 순환 종양 RNA(circulating tumor RNA, ctRNA)나 엑소좀(exosome)에 포함된 RNA를 분석하여 암을 조기에 진단하거나 치료 반응을 모니터링하려는 연구가 활발히 진행되고 있으며, 이러한 미량의 RNA를 검출하고 분석하는 데 고감도 RT-PCR 기술이 필수적으로 요구됩니다 [22].
기타 응용 분야
위에 소개된 주요 분야 외에도 RT-PCR 기술은 다양한 생명 과학 연구 분야에서 폭넓게 활용되고 있습니다. 예를 들어, RNA 간섭(RNA interference, RNAi) 연구에서는 siRNA 등을 이용하여 특정 유전자의 mRNA를 인위적으로 조절한 후, 그 효과를 검증하기 위해 표적 mRNA 발현량 감소 여부를 RT-qPCR로 확인합니다.
또한, 특정 세포나 조직에서 발현되는 전체 mRNA 집단을 연구하기 위한 cDNA 라이브러리 제작이나, 특정 유전자의 기능을 상세히 연구하거나 재조합 단백질을 생산하기 위한 유전자 클로닝 과정에서도 역전사 단계가 필수적으로 사용됩니다. 최근에는 단백질을 만들지는 않지만 유전자 발현 조절 등에 중요한 역할을 하는 MicroRNA(miRNA) 및 기타 비번역 RNA(non-coding RNA) 연구에서도 특화된 RT-PCR 방법들이 개발되어 그 발현 양상을 분석하는 데 활발히 활용되고 있습니다.
이처럼 RT-PCR은 RNA가 관여하는 거의 모든 생명 현상을 연구하고 이해하는 데 없어서는 안 될 강력하고 다재다능한 기술이라고 할 수 있습니다.
RT-PCR의 장점과 한계점
RT-PCR은 의심할 여지 없이 RNA 분석에 있어 혁명적인 발전을 가져온 매우 강력하고 유용한 도구입니다. 하지만 모든 기술이 그렇듯이 완벽하지만은 않으며, 몇 가지 내재적인 한계점과 사용 시 주의해야 할 점들을 가지고 있습니다. 따라서 RT-PCR 기술을 올바르게 이해하고 효과적으로 활용하기 위해서는 그 장점과 단점을 명확히 아는 것이 중요합니다. 마치 뛰어난 성능의 자동차라도 그 특성과 조작 시 주의사항을 알아야 안전하고 효율적으로 운전할 수 있는 것과 같습니다.
RT-PCR의 장점
RT-PCR 기술이 광범위하게 사용되는 이유는 여러 명확한 장점들 때문입니다. 타의 추종을 불허하는 민감도(High Sensitivity)는 RT-PCR의 가장 큰 장점으로, PCR 과정의 기하급수적인 증폭 능력 덕분에 극미량의 RNA 분자도 검출할 수 있습니다. 이는 감염 초기 바이러스 진단, 희귀 유전자 전사체 연구, 미량 시료 분석 등에서 엄청난 위력을 발휘합니다.
또한, 특정 염기서열에만 결합하도록 세심하게 설계된 프라이머를 사용하기 때문에 표적을 놓치지 않는 높은 특이도(High Specificity)를 자랑합니다. 복잡한 RNA 혼합물 속에서도 목표하는 RNA 유래 cDNA만을 선택적으로 증폭할 수 있으며, 특히 TaqMan 프로브와 같은 서열 특이적인 형광 프로브를 사용하는 RT-qPCR은 특이도를 더욱 향상시킵니다.
더불어 정량 분석의 가능성(Quantification Capability) 또한 RT-PCR의 중요한 장점입니다. 특히 RT-qPCR 기술을 이용하면 단순히 RNA의 존재 유무를 넘어 초기 시료에 존재했던 RNA의 양을 정확하게 정량화할 수 있습니다. Ct 값이라는 객관적인 지표를 통해 유전자 발현 수준의 미세한 변화를 비교 분석하거나 바이러스 부하 등을 측정하는 것이 가능합니다.
이러한 특성 덕분에 RT-PCR은 넓은 응용 범위(Wide Applicability)를 가집니다. 기초 생명 과학 연구부터 임상 진단, 신약 개발, 법의학, 식품 안전, 환경 모니터링 등 RNA가 관여하는 거의 모든 영역에서 활용될 수 있습니다. 마지막으로, 특히 One-step RT-PCR이나 자동화 시스템을 이용하면 상대적으로 빠른 분석 시간(Relatively Fast Turnaround Time) 내에 결과를 얻을 수 있어 신속한 진단이 중요한 감염병 상황 등에서 큰 이점을 제공합니다.
RT-PCR의 한계점 및 주의사항
이러한 강력한 장점들에도 불구하고, RT-PCR 실험을 수행하고 결과를 해석할 때는 몇 가지 한계점과 잠재적인 문제점들을 반드시 고려해야 합니다. 가장 근본적인 문제점은 RNA의 불안정성과 RNase 오염의 위협입니다. RNA는 DNA보다 화학적으로 훨씬 불안정하며, 어디에나 존재하는 RNase에 의해 매우 쉽게 분해되므로, 시료 처리 및 실험 전 과정에서 RNA 분해 방지와 RNase 오염 방지를 위한 각별한 주의가 필요합니다. 이는 실험의 성패를 좌우하는 가장 중요하고도 어려운 부분으로, 제대로 관리되지 않으면 위음성 결과나 부정확한 정량 결과를 초래할 수 있습니다.
또한, 역전사 효율의 변동성(Variability in RT Efficiency)도 중요한 고려사항입니다. RNA를 cDNA로 변환하는 역전사 단계의 효율은 100%가 아니며, 사용하는 효소, 프라이머, RNA 품질, 반응 조건, 저해 물질 존재 여부 등 여러 요인에 의해 영향을 받습니다 [12].
이러한 변동성은 특히 여러 샘플 간 RNA 양을 비교하는 상대 정량 분석 결과의 정확성에 영향을 줄 수 있으므로, 실험 조건을 최대한 일정하게 유지하고 신뢰성 있는 시약 사용 및 적절한 대조군 설정이 중요합니다. 더불어, 시료 자체에 PCR 저해 물질(Presence of PCR Inhibitors)이 포함되어 있을 가능성도 있습니다. 혈액의 헤모글로빈이나 조직의 콜라겐, 추출 과정에서 잔류하는 유기 용매나 염 등이 효소 활성을 억제하여 PCR 효율을 떨어뜨릴 수 있으므로 [23], 효과적인 정제 과정을 통해 저해 물질을 최대한 제거하고 필요 시 내부 대조군을 사용해 저해 여부를 모니터링해야 합니다.
게놈 DNA(gDNA) 오염의 함정 역시 주의해야 합니다. RNA 추출 시 소량의 gDNA가 함께 오염될 수 있는데, 만약 프라이머가 gDNA에도 결합하여 증폭이 일어난다면 실제 mRNA 발현량을 과대평가하는 위양성 결과를 초래할 수 있습니다. 이를 방지하기 위해 DNase 처리, 인트론-스패닝 프라이머 설계, 또는 역전사 효소를 넣지 않은 RT-미처리 대조군(-RT control) 포함 등의 전략을 사용해야 합니다.
정량 분석 측면에서는, 겔 전기영동으로 결과를 확인하는 Endpoint RT-PCR의 경우 정확한 정량의 어려움이 있습니다. PCR 반응 후반부의 포화 효과 때문에 초기 주형 양의 차이가 최종 산물 양에 제대로 반영되지 않을 수 있으므로, 정밀한 정량이 필요하다면 반드시 RT-qPCR을 사용해야 합니다.
마지막으로, RT-qPCR 데이터의 결과 해석의 복잡성도 인지해야 합니다. 상대 정량 분석 시 샘플 간 차이를 보정하기 위한 참조 유전자(Reference Gene)의 선택과 검증이 매우 중요하며 [13, 24], 잘못된 참조 유전자 사용은 결과 해석에 심각한 오류를 야기할 수 있습니다. 또한, PCR 증폭 효율이 유전자마다 다를 수 있으므로 이를 고려하여 데이터를 보정해야 할 수 있으며, 적절한 대조군 설정과 통계 처리를 통해 결과의 신뢰성과 유의성을 확보해야 합니다.
아래 표는 RT-PCR의 장점과 한계점을 다시 한번 요약 정리한 것입니다.
| 장점 | 한계점 및 고려사항 |
|---|---|
| 높은 민감도 | RNA 불안정성 및 RNase 오염 민감성 |
| 높은 특이도 | 역전사 효율의 변동성 |
| 정량 분석 가능 (RT-qPCR) | PCR 저해 물질 존재 가능성 |
| 넓은 응용 범위 | 게놈 DNA (gDNA) 오염 가능성 |
| 상대적으로 빠른 분석 시간 | 정확한 정량의 어려움 (Endpoint) |
| 결과 해석의 복잡성 (참조 유전자, 효율 등) |
이처럼 RT-PCR은 강력한 분석 도구이지만, 그 결과를 맹신하기보다는 실험 과정의 각 단계별 특성과 잠재적 오류 가능성을 충분히 인지하고, 엄격한 품질 관리와 적절한 데이터 해석을 통해 신뢰성 있는 결론을 도출하려는 노력이 반드시 필요합니다.
결론 RT-PCR의 현재와 미래
자, 지금까지 우리는 역전사 중합효소연쇄반응(RT-PCR)이라는 기술에 대해 아주 깊이 있게 탐험해 보았습니다. RNA 정보를 DNA로 번역하는 '역전사' 과정과 번역된 DNA 정보를 수백만 배로 증폭하는 'PCR' 과정의 절묘한 만남이 바로 RT-PCR의 핵심 원리라는 것을 이해하셨을 겁니다. 불안정하고 직접 다루기 힘든 RNA를 분석 가능한 형태로 바꾸고, 그 양을 민감하게 측정할 수 있게 해주는 이 기술의 강력함은 실로 대단하다고 할 수 있지요.
우리는 또한 실험실에서 RT-PCR이 어떻게 수행되는지, 그 단계별 과정과 주의사항들을 상세히 살펴보았습니다. 특히 모든 단계에서 RNase 오염을 막는 것이 얼마나 중요한지, 그리고 실험 목적에 따라 Two-step과 One-step RT-PCR, Endpoint RT-PCR과 RT-qPCR 중 어떤 방법을 선택해야 하는지에 대해서도 알아보았습니다. 고품질 RNA 확보부터 신중한 프라이머 설계, 반응 조건 최적화, 그리고 정확한 결과 분석에 이르기까지, 신뢰성 있는 결과를 얻기 위한 여정은 결코 간단하지 않다는 것도 느끼셨을 겁니다.
하지만 이러한 노력 덕분에 RT-PCR 기술은 유전자 발현 연구를 통해 생명 현상의 비밀을 밝히고, 감염성 질환 진단의 최전선에서 활약하며(COVID-19 팬데믹 극복에 기여했듯이!), 유전 질환과 암의 원인을 규명하고 진단하는 데 결정적인 역할을 수행하고 있습니다. 그야말로 현대 생명 과학과 의학 연구를 떠받치는 기둥과 같은 기술이라고 할 수 있습니다.
물론, RNA의 불안정성, 역전사 효율의 변동성, gDNA 오염 가능성, 결과 해석의 복잡성과 같은 기술적인 한계점과 도전 과제들도 분명히 존재합니다. 따라서 우리는 RT-PCR 결과를 해석할 때 항상 비판적인 시각을 유지하고, 실험 설계와 수행, 데이터 분석 과정에서 발생할 수 있는 오류들을 최소화하기 위해 끊임없이 노력해야 합니다.
앞으로 RT-PCR 기술은 더욱 발전해 나갈 것입니다. 한 분자 수준에서 RNA를 절대 정량할 수 있는 디지털 PCR(Digital PCR) 기술과의 융합, 시료 처리부터 결과 분석까지 전 과정을 자동화하는 시스템의 발전, 더 높은 효율과 안정성을 가진 효소 및 시약의 개발 등은 RT-PCR 기술을 더욱 민감하고, 정확하며, 빠르고, 간편하게 만들어 줄 것입니다. 이러한 기술의 진보는 정밀 의학(precision medicine) 시대를 앞당기고, 질병의 조기 진단과 개인 맞춤형 치료 전략 개발에 크게 기여할 것으로 기대됩니다.
2025.04.13 - [인증심사 및 검사실 운영] - 검사실 위험관리 기법 CLSI EP18-A2 (구성요소, 위험사정도구, 위험관리 영역, 인증심사 가이드)
검사실 위험관리 기법 CLSI EP18-A2 (구성요소, 위험사정도구, 위험관리 영역, 인증심사 가이드)
안녕하세요? 여러분은 혹시 병원에서 검사를 받고 그 결과를 기다리면서, '이 검사 결과가 정말 정확할까?' 하는 생각을 해보신 적이 있으신가요? 매일 수많은 검사가 이루어지는 임상 검사실에
labdoctor.tistory.com
2025.04.13 - [분자진단] - FISH (Flourescence in situ hybridization) 의 원리와 방법, 적용 분야
FISH (Flourescence in situ hybridization) 의 원리와 방법, 적용 분야
우리 몸의 세포 하나하나에는 생명의 모든 정보가 담긴 거대한 도서관, 즉 유전체가 존재합니다. 이 도서관에는 수많은 책(염색체)이 있고, 각 책에는 무수히 많은 이야기(유전자)가 적혀 있습니
labdoctor.tistory.com
2025.04.13 - [분자진단] - NGS (Next Generation Sequencing)의 개념, 원리, 방법, 적용 분야
NGS (Next Generation Sequencing)의 개념, 원리, 방법, 적용 분야
이번 시간에는 현대 생명과학과 의학 연구의 패러다임을 바꾼 혁신적인 기술, 차세대 염기서열 분석(Next Generation Sequencing, NGS)에 대해 심도 있게 알아보겠습니다. 혹시 몇 년 전, 유명 할리우드
labdoctor.tistory.com
2025.04.13 - [분자진단] - SNP (Single Nucleotide Polymorphism)의 정의, 임상적 의의, 검사 방법
SNP (Single Nucleotide Polymorphism)의 정의, 임상적 의의, 검사 방법
우리 인간은 모두 같은 '사람'이라는 종에 속하지만, 놀랍도록 다채로운 모습과 특성을 지니고 살아갑니다. 쌍둥이가 아닌 이상, 세상에 외모부터 성격, 심지어 특정 질병에 걸릴 확률이나 약물
labdoctor.tistory.com
참고문헌
[1] Baltimore, D. (1970). RNA-dependent DNA polymerase in virions of RNA tumour viruses. Nature, 226(5252), 1209-1211.
[2] Mullis, K. B., & Faloona, F. A. (1987). Specific synthesis of DNA in vitro via a polymerase-catalyzed chain reaction. Methods in Enzymology, 155, 335-350.
[3] Sambrook, J., & Russell, D. W. (2001). Molecular Cloning: A Laboratory Manual (3rd ed.). Cold Spring Harbor Laboratory Press. (Chapter 7: Extraction, Purification, and Analysis of mRNA from Eukaryotic Cells)
[4] Manchester, K. L. (1996). Use of UV spectroscopy for determination of protein or nucleic acid concentration. BioTechniques, 20(6), 968-970.
[5] Rychlik, W. (1995). Primer selection for PCR. Molecular Biotechnology, 3(2), 129-134.
[6] Bustin, S. A., Benes, V., Garson, J. A., Hellemans, J., Huggett, J., Kubista, M., ... & Wittwer, C. T. (2009). The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clinical Chemistry, 55(4), 611-622.
[7] Pfaffl, M. W. (2001). A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Research, 29(9), e45.
[8] Corman, V. M., Landt, O., Kaiser, M., Molenkamp, R., Meijer, A., Chu, D. K., ... & Drosten, C. (2020). Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Eurosurveillance, 25(3), 2000045.
[9] Wang, G. S., & Cooper, T. A. (2007). Splicing in disease: disruption of the splicing code and the decoding machinery. Nature Reviews Genetics, 8(10), 749-761.
[10] Slamon, D. J., Leyland-Jones, B., Shak, S., Fuchs, H., Paton, V., Bajamonde, A., ... & Press, M. F. (2001). Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. New England Journal of Medicine, 344(11), 783-792.
[11] Hughes, T. P., Deininger, M., Hochhaus, A., Branford, S., Radich, J., Kaeda, J., ... & Goldman, J. M. (2006). Monitoring CML patients responding to treatment with tyrosine kinase inhibitors: review and recommendations for harmonizing current methodology for detecting BCR-ABL transcripts and kinase domain mutations and for expressing results. Blood, 108(1), 28-37.
[12] Stahlberg, A., Håkansson, J., Xian, X., Semb, H., & Kubista, M. (2004). Properties of the reverse transcription reaction in mRNA quantification. Clinical Chemistry, 50(3), 509-515.
[13] Vandesompele, J., De Preter, K., Pattyn, F., Poppe, B., Van Roy, N., De Paepe, A., & Speleman, F. (2002). Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biology, 3(7), research0034.
[14] Temin, H. M., & Mizutani, S. (1970). RNA-dependent DNA polymerase in virions of Rous sarcoma virus. Nature, 226(5252), 1211-1213.
[15] Gerard, G. F., & D'Alessio, J. M. (1993). Reverse transcriptase: the use of cloned Moloney murine leukemia virus reverse transcriptase to synthesize DNA from RNA. Methods in Molecular Biology, 16, 73-93.
[16] Lanier, E. R., & D'Aquila, R. T. (1997). Comparison of random-primed versus oligo(dT)-primed cDNA synthesis for Measuring HIV-1 RNA levels. Journal of Virological Methods, 65(1), 57-68.
[17] Freeman, W. M., Walker, S. J., & Vrana, K. E. (1999). Quantitative RT-PCR: pitfalls and potential. BioTechniques, 26(1), 112-125.
[18] Chomczynski, P., & Sacchi, N. (1987). Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Analytical Biochemistry, 162(1), 156-159.
[19] Schroeder, A., Mueller, O., Stocker, S., Salowsky, R., Leiber, M., Gassmann, M., ... & Hubber, A. (2006). The RIN: an RNA integrity number for assigning integrity values to RNA measurements. BMC Molecular Biology, 7(1), 3.
[20] Dieffenbach, C. W., Lowe, T. M., & Dveksler, G. S. (1993). General concepts for PCR primer design. PCR Methods and Applications, 3(3), S30-S37.
[21] Finkel, R. S., Mercuri, E., Darras, B. T., Connolly, A. M., Kuntz, N. L., Kirschner, J., ... & Bertini, E. (2017). Nusinersen versus sham control in infantile-onset spinal muscular atrophy. New England Journal of Medicine, 377(18), 1723-1732.
[22] Schwarzenbach, H., Hoon, D. S., & Pantel, K. (2011). Cell-free nucleic acids as biomarkers in cancer patients. Nature Reviews Cancer, 11(6), 426-437.
[23] Wilson, I. G. (1997). Inhibition and facilitation of nucleic acid amplification. Applied and Environmental Microbiology, 63(10), 3741-3751.
[24] Huggett, J., Dheda, K., Bustin, S., & Zumla, A. (2005). Real-time RT-PCR normalisation; strategies and considerations. Genes and Immunity, 6(4), 279-284.
'분자진단' 카테고리의 다른 글
| 염기서열분석의 역사와 종류, 특징, 차이점 (0) | 2025.04.13 |
|---|---|
| Pyrosequencing 의 원리와 방법, 적용 분야 (0) | 2025.04.13 |
| FISH (Flourescence in situ hybridization) 의 원리와 방법, 적용 분야 (1) | 2025.04.13 |
| NGS (Next Generation Sequencing)의 개념, 원리, 방법, 적용 분야 (0) | 2025.04.13 |
| SNP (Single Nucleotide Polymorphism)의 정의, 임상적 의의, 검사 방법 (0) | 2025.04.13 |




댓글