진단검사의학 분야에서 정확하고 신뢰할 수 있는 검사 결과를 제공하는 것은 환자 진료의 핵심입니다. 만약 혈당 검사 결과가 실제보다 낮게 나온다면 어떻게 될까요? 의사는 이를 바탕으로 잘못된 치료 결정을 내릴 수 있고, 이는 환자에게 심각한 결과를 초래할 수 있습니다. 이러한 불상사를 막기 위해 검사실에서는 사용하는 시약이나 장비가 일관된 성능을 유지하도록 엄격하게 관리해야 하는데, 특히 검사에 사용하는 시약은 제조 과정상의 미세한 차이로 인해 로트(lot, 동일 조건에서 생산된 제품의 묶음 단위)마다 성능이 조금씩 다를 수 있습니다 [13, 24].
이 때문에 검사실에서는 새로운 로트의 시약을 사용하기 전에 반드시 기존 로트와의 성능 차이가 임상적으로 허용 가능한 범위 내에 있는지를 평가하는 과정을 거쳐야 합니다. 이를 시약 로트 변경 검증(reagent lot change verification) 또는 로트 간 검증(lot-to-lot verification, LTLV)이라고 부릅니다 [4, 7, 13]. 마치 새로운 요리 재료를 사용하기 전에 맛이 기존과 크게 다르지 않은지 확인하는 것과 비슷하다고 할 수 있습니다.
하지만 이 로트 변경 검증 과정은 생각보다 복잡하고 시간과 자원이 많이 소요될 수 있습니다 [7, 12, 15]. 특히 로트 변경이 잦은 검사실에서는 이 과정이 상당한 부담으로 작용할 수 있습니다. 그래서 임상검사실표준협회(Clinical and Laboratory Standards Institute, CLSI)에서는 EP26-Ed2라는 지침을 통해 이 과정을 간소화하면서도 통계적으로 타당한 방법을 제시하고 있습니다 [12, 15, 16, 27]. 이 지침의 핵심은 바로 임계 차이(Critical Difference, CD)'라는 개념을 활용하여 최소한의 검체만으로도 로트 간의 유의미한 차이를 효과적으로 평가하는 것입니다 [12, 15, 17].
이번 시간에는 바로 이 CLSI EP26-Ed2 지침에서 제시하는 간소화된 로트 변경 검증 방법에 대해, 배경지식이 없는 독자도 쉽게 이해할 수 있도록 기초 개념부터 시작하여 깊이 있는 수준까지 상세하게 알아보겠습니다
시약 로트 변경 검증 중요성 및 기본 개념
로트 변경 검증 필요성
검사실에서 새로운 시약 로트를 도입할 때 왜 반드시 검증 절차를 거쳐야 할까요? 얼핏 생각하면 같은 제조사에서 만든 동일한 제품인데 굳이 번거로운 검증까지 필요할까 싶을 수도 있습니다. 하지만 이는 환자 안전과 진료의 질에 직결되는 매우 중요한 문제입니다 [2, 10].
시약은 화학 물질의 복잡한 혼합물이며, 제조 공정 중에는 원재료의 미세한 차이, 생산 환경의 변화, 보관 및 운송 조건 등 수많은 변수가 작용할 수 있습니다 [7]. 이러한 변수들은 겉보기에는 동일해 보이는 시약이라도 로트별로 미묘한 성능 차이를 유발할 수 있습니다. 예를 들어, 특정 효소 활성도가 약간 다르거나, 항체의 결합력이 미세하게 변하거나, 반응 속도가 조금 달라지는 등의 차이가 발생할 수 있다는 것입니다.
만약 이러한 성능 차이를 인지하지 못하고 새로운 로트의 시약을 그대로 사용한다면 어떻게 될까요? 검사 결과값이 이전 로트와 달라질 수 있습니다. 즉, 동일한 환자의 검체를 측정하더라도 사용하는 시약 로트에 따라 다른 결과값이 나올 수 있다는 의미입니다. 이러한 결과의 비일관성'은 의료진에게 혼란을 야기하고, 환자의 상태를 잘못 판단하게 만들 수 있습니다 [7, 10].
예를 들어, 특정 질병 표지자 검사에서 새로운 로트 시약이 이전보다 약간 낮은 값을 내는 경향이 있다면, 실제로는 질병이 진행 중인 환자를 정상으로 오인할 위험이 있습니다. 반대로 약간 높은 값을 내는 경향이 있다면, 건강한 사람을 불필요하게 추가 검사나 치료 대상으로 분류할 수도 있습니다. 이는 환자에게 잘못된 진단, 부적절한 치료, 불필요한 불안감과 의료비용 부담을 안겨줄 수 있는 심각한 문제입니다 [2, 5].
품질관리(QC) 물질을 측정하는 것만으로는 이러한 문제를 완벽하게 예방하기 어렵습니다 [13, 27]. 왜냐하면 QC 물질은 실제 환자 검체와 그 성상(matrix)이 다를 수 있기 때문입니다. 따라서 QC 결과에서는 별다른 차이가 없었더라도, 실제 환자 검체에서는 유의미한 차이가 나타날 수 있습니다 [27]. 마치 특정 음식 알레르기가 있는 사람에게는 괜찮았던 재료가, 다른 사람에게는 심각한 알레르리 반응을 일으킬 수 있는 것과 유사합니다. QC 물질은 일종의 '표준 샘플'과 같아서, 실제 환자 검체에서 발생할 수 있는 다양한 변이를 모두 반영하지 못할 수 있다는 것입니다.
따라서 새로운 시약 로트를 사용하기 전에는 반드시 실제 환자 검체를 이용하여 기존 로트와 비교 평가를 수행하고, 그 차이가 임상적으로 허용 가능한 수준인지를 확인하는 로트 변경 검증 절차가 필수적입니다 [7, 13, 16, 27]. 이는 마치 새로운 약을 시판하기 전에 임상시험을 통해 안전성과 유효성을 검증하는 것과 같은 이치입니다. 검사실의 이러한 노력은 검사 결과의 일관성과 신뢰성을 보장하고, 궁극적으로 환자 안전을 지키는 데 핵심적인 역할을 수행합니다 [2, 10, 13]. 또한, 많은 인증 기관에서도 로트 변경 검증을 필수 요건으로 요구하고 있습니다 [5, 7, 32].
검사 결과 변동성 및 비정밀도 이해
CLSI EP26 지침의 핵심 원리를 이해하기 위해서는 먼저 검사실 검사 결과에 내재된 '변동성', 즉 비정밀도(imprecision)에 대한 이해가 필수적입니다. 왜냐하면 EP26 방법은 바로 이 비정밀도와 비교하여 로트 간 차이의 수용 가능성을 판단하기 때문입니다 [12, 15].
세상에 완벽하게 동일한 측정값을 반복적으로 내는 측정 시스템은 존재하지 않습니다. 아무리 정교한 장비와 숙련된 검사자라 할지라도, 동일한 검체를 여러 번 반복 측정하면 결과값은 미세하게 달라지게 마련입니다. 이러한 측정값들의 흩어짐 정도, 즉 결과의 재현성(reproducibility)을 나타내는 지표가 바로 정밀도(precision)입니다 [5, 6, 10]. 정밀도가 높다는 것은 반복 측정 시 결과값들이 서로 가깝게 모여 있다는 의미이며, 반대로 정밀도가 낮다는 것은 결과값들이 넓게 흩어져 있다는 의미입니다. 흔히 비정밀도(imprecision)라는 용어는 정밀도가 낮은 정도, 즉 결과값의 변동성(variability) 자체를 의미하며, 정밀도와는 반대 개념으로 사용됩니다 [3, 5, 10].
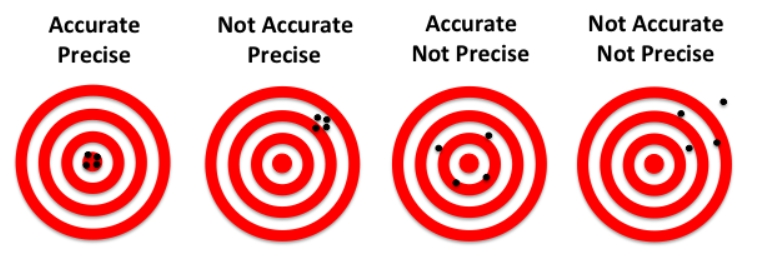
위 이미지는 정확도(accuracy, 참값에 얼마나 가까운가)와 정밀도(precision, 반복 측정 시 결과가 얼마나 모여 있는가)의 개념을 보여줍니다. 높은 정밀도는 반복 측정 결과가 서로 가깝게 모여 있는 상태를 의미합니다.
그렇다면 이러한 비정밀도는 왜 발생하는 것일까요? 여기에는 다양한 요인이 복합적으로 작용합니다 [6, 28]. 분석 시스템 내부의 미세한 온도 변화, 시약 공급 펌프의 미세한 유량 변동, 광원 강도의 미세한 흔들림 등 기계적인 불안정성이나, 동일 로트 내에서도 시약 병마다의 미세한 농도 차이, 시약 분주 과정의 오차, 시약의 점진적인 변성 등 시약 관련 요인 때문일 수 있습니다.
또한 검체를 다루거나 장비를 조작하는 과정에서의 미세한 습관 차이, 수동 단계에서의 미세한 오차 등 검사자 관련 요인이나 검사실의 온도, 습도, 진동 등의 미세한 변화와 같은 환경 요인도 비정밀도에 영향을 미칩니다. 이러한 요인들이 복합적으로 작용하여 측정 결과에 무작위적인 변동(random error)을 유발하며, 이것이 바로 비정밀도로 나타나는 것입니다 [5, 6, 10].
비정밀도는 통계적으로 표준편차(Standard Deviation, SD)나 변이계수(Coefficient of Variation, CV)로 정량화됩니다 [3, 5]. 표준편차는 결과값들이 평균값으로부터 얼마나 떨어져 있는지를 나타내는 절대적인 값이고, 변이계수는 표준편차를 평균값으로 나눈 상대적인 값(%CV)으로, 측정값의 크기에 관계없이 변동성을 비교할 때 유용하게 사용됩니다 [5, 6, 28]. 즉, %CV가 작을수록 정밀도가 높다는 의미입니다.
CLSI 지침에서는 비정밀도를 측정 조건에 따라 다음과 같이 세분화하기도 합니다 [6, 9, 28, 29]. 먼저 반복 정밀도 (Repeatability, 이전의 'Within-run precision', Sr 또는 CVr)는 가장 짧은 시간 간격 내에서, 동일한 검사자, 동일한 장비, 동일한 시약 로트, 동일한 작동 조건 하에서 동일 검체를 반복 측정했을 때의 비정밀도를 의미합니다 [6, 9, 28, 29]. 이는 주로 분석 시스템 자체의 단기적인 무작위 변동성을 반영합니다.
다음으로 검사실 내 정밀도 (Within-laboratory precision, 이전의 'Total precision', SwL 또는 CVwL)는 장기간(예: 수 주 또는 수 개월)에 걸쳐, 동일한 검사실 내에서, 다른 검사자, 다른 시약 로트(통상적으로는 동일 로트 내 다른 바이알), 다른 보정(calibration) 주기 등 일상적인 운영 조건 하에서 발생하는 비정밀도를 모두 포함합니다 [6, 9, 28, 29]. 이는 반복 정밀도에 더해 시간 경과에 따른 변동 요인(예: 장비 노후화, 환경 변화, 검사자 간 차이 등)까지 반영하는, 보다 현실적인 검사실 운영 환경에서의 총 비정밀도를 나타냅니다. CLSI EP26 지침에서 로트 변경 검증 시 주로 고려하는 비정밀도가 바로 이 검사실 내 비정밀도(CVwL)입니다 [12, 15, 17, 21].
마지막으로 재현성 (Reproducibility, Sr 또는 CVr)은 서로 다른 검사실, 다른 장비, 다른 검사자 등 매우 다른 조건 하에서 동일 검체를 측정했을 때의 비정밀도를 의미합니다 [29]. 이는 검사 방법 자체의 견고성(robustness)을 평가하는 데 사용됩니다. 이러한 비정밀도의 개념, 특히 검사실 내 비정밀도(CVwL)를 이해하는 것은 CLSI EP26의 간소화된 방법을 파악하는 데 있어 첫걸음이라고 할 수 있습니다. 왜냐하면 바로 이 CVwL 값이 로트 간 차이의 허용 기준인 임계 차이(CD)와 비교되는 핵심적인 '잡음(noise)' 수준을 나타내기 때문입니다.
임상적 허용 기준 임계 차이 (CD)
CLSI EP26 지침의 또 다른 핵심 개념은 바로 임계 차이(Critical Difference, CD)입니다. 우리말로 '결정적인 차이', '중요한 차이' 정도로 해석할 수 있겠는데요, 이는 새로운 시약 로트와 기존 시약 로트 간에 발생할 수 있는 결과 차이 중, 임상적으로 수용 가능한 최대 허용 한계를 의미합니다 [7, 12, 13, 15, 21]. 쉽게 말해, "이 정도 차이까지는 환자 진료에 큰 영향을 미치지 않으니 괜찮다고 보자"라고 검사실이 미리 설정하는 기준값인 것입니다 [13].
아니, 그냥 통계적으로 유의미한 차이가 없으면 되는 거 아냐? 왜 굳이 '임계 차이'라는 걸 따로 정해야 해? 아주 좋은 질문입니다! 많은 분들이 이렇게 생각하실 수 있습니다. 하지만 통계적 유의성(statistical significance)과 임상적 유의성(clinical significance)은 다른 개념이라는 점을 반드시 기억해야 합니다. 통계적으로 유의하다는 것은 단순히 두 그룹 간의 차이가 우연히 발생했을 확률이 매우 낮다는 것을 의미할 뿐, 그 차이가 실제 환자 진료에 의미 있는 영향을 미치는지는 말해주지 않습니다 [2].
예를 들어, 어떤 혈당 검사의 새로운 시약 로트가 기존 로트보다 평균적으로 0.1 mg/dL 높은 결과를 보인다고 가정해 봅시다. 만약 검사를 매우 많이 반복하여 정밀도가 극도로 높다면, 이 0.1 mg/dL라는 작은 차이도 통계적으로는 '유의하다'고 나올 수 있습니다. 하지만 혈당 0.1 mg/dL 차이는 환자의 당뇨 진단이나 치료 방침 결정에 아무런 영향도 미치지 못하는, 임상적으로는 전혀 중요하지 않은 차이입니다. 이런 경우까지 로트 변경을 거부한다면 불필요한 자원 낭비만 초래할 뿐입니다.
반대로, 어떤 검사에서는 통계적으로는 유의하지 않더라도, 그 차이가 환자에게 심각한 영향을 줄 수 있는 경우도 있습니다. 예를 들어, 치료 약물 농도 모니터링 검사에서 로트 간 차이가 통계적으로는 유의하지 않게 나왔지만, 그 차이가 허용 범위를 약간 벗어나 약물 용량 조절에 영향을 미칠 수 있다면 이는 임상적으로 매우 중요한 문제가 될 수 있습니다.
바로 이러한 이유 때문에 CLSI EP26에서는 단순히 통계적 유의성만을 따지는 것이 아니라, 임상적으로 의미 있는 차이'의 기준, 즉 임계 차이(CD)를 설정하도록 요구하는 것입니다 [7, 13, 16]. CD는 각 검사 항목의 특성과 임상적 중요성을 고려하여 설정되어야 합니다. 예를 들어, 치료 범위가 좁은 약물 농도 검사나 생리적 변동이 매우 작은 전해질 검사 등에서는 작은 차이도 임상적으로 중요할 수 있으므로 CD를 작게 설정해야 합니다. 반면, 생리적 변동 자체가 매우 크거나 임상적 판단에 여유가 있는 검사 항목의 경우 CD를 상대적으로 크게 설정할 수 있습니다.
그렇다면 이 중요한 임계 차이(CD)는 어떻게 결정할까요? CLSI EP26 지침은 CD를 설정하는 구체적인 방법을 명시하기보다는, 각 검사실이 해당 검사 항목의 임상적 활용도(clinical use)를 바탕으로 결정해야 한다고 권고합니다 [13]. 하지만 일반적으로 총 허용 오차(Total Allowable Error, TEa) 개념에 기반한 허용 바이어스(allowable bias) 목표치를 CD 설정에 활용할 수 있습니다 [12, 14, 15].
총 허용 오차(Total Allowable Error, TEa)란, 검사 결과의 부정확함(bias, 계통 오차)과 비정밀도(imprecision, 무작위 오차)를 모두 포함하여, 해당 검사 결과가 임상적으로 유용하기 위해 허용될 수 있는 최대 오차의 한계를 의미합니다 [1, 3, 4, 5, 10, 14]. 즉, "이 검사 결과는 참값에서 이 정도(TEa)까지 벗어나도 임상적 판단에 영향을 주지 않는다는" 기준입니다 [3, 4, 10].
TEa는 크게 두 가지 요소, 즉 계통 오차(Systematic Error)를 나타내는 바이어스(Bias)'와 무작위 오차(Random Error)를 나타내는 비정밀도(Imprecision)'로 구성됩니다 [3, 5, 10]. 바이어스는 측정값이 참값에서 일관되게 벗어나는 경향, 즉 정확도(Accuracy)의 부족을 의미하고 [1, 2, 28], 비정밀도는 반복 측정 시 결과값의 흩어짐, 즉 정밀도(Precision)의 부족을 의미합니다 [1, 2, 28].
시약 로트 변경으로 인해 발생하는 주된 문제는 일반적으로 계통 오차, 즉 바이어스(bias)'의 변화입니다 [1, 2, 3]. 물론 비정밀도에도 영향을 줄 수 있지만, 새로운 로트가 이전 로트와 비교하여 결과값을 평균적으로 더 높게 또는 더 낮게 내는 경향, 즉 결과값의 평균적인 이동(shift)이 주요 관심사라는 것입니다. 따라서 로트 변경 검증의 허용 기준인 CD를 설정할 때, TEa 구성 요소 중 허용 바이어스(Allowable Bias) 부분을 기준으로 삼는 것이 논리적으로 타당합니다 [12, 14, 15].
그렇다면 '허용 바이어스'는 어떻게 결정될까요? 이는 TEa를 어떻게 정의하느냐에 따라 달라집니다. 다양한 TEa 모델이 있지만, 생물학적 변동(Biological Variation, BV)에 기반한 모델이 가장 널리 사용되는 방법 중 하나입니다 [4, 10, 14, 15]. 생물학적 변동 데이터베이스(예: EFLM Biological Variation Database)는 수많은 검사 항목에 대해 개인 내 변동(CVi)과 개인 간 변동(CVg) 값을 제공합니다. 이를 바탕으로 분석 성능 목표(Analytical Performance Specifications, APS)를 설정할 수 있는데, 일반적으로 다음과 같은 세 가지 수준의 목표를 제시합니다 [1, 2, 14].
최소 성능 (Minimum performance)입니다. 허용 바이어스는 0.375 × (CVi² + CVg²)^(1/2) 이하, 허용 CVa는 0.75 × CVi 이하입니다. 여기서 CVa는 분석적 비정밀도, 즉 검사 자체의 CV를 의미합니다.
바람직한 성능 (Desirable performance)입니다. 허용 바이어스는 0.250 × (CVi² + CVg²)^(1/2) 이하, 허용 CVa는 0.50 × CVi 이하입니다.
최적 성능 (Optimum performance)입니다. 허용 바이어스는 0.125 × (CVi² + CVg²)^(1/2) 이하, 허용 CVa는 0.25 × CVi 이하입니다.
검사실은 자신들의 품질 목표 수준(최소, 바람직한, 최적)에 맞춰 위 공식 등을 이용하여 각 검사 항목별 허용 바이어스 목표치를 계산할 수 있습니다. 이렇게 계산된 허용 바이어스 값을 CD로 사용할 수 있는 것입니다 [14, 15].
다른 방법으로는 CLIA'88과 같은 규제 기관의 기준을 활용하는 것입니다. CLIA'88은 외부정도관리(Proficiency Testing, PT) 프로그램에서 합격/불합격 판정을 위한 허용 오차 범위를 규정하고 있습니다 [4, 5, 7]. 이 기준은 종종 TEa 목표치로 간주되며, 이 기준의 일부(예: TEa의 1/2, 1/3 등)를 허용 바이어스, 즉 CD로 설정하기도 합니다 [14, 17]. 예를 들어, CLIA에서 포도당(Glucose)의 TEa를 ±6 mg/dL 또는 ±10% 중 큰 값으로 규정하고 있다면, 검사실은 CD를 이 값의 절반인 ±3 mg/dL 또는 ±5%로 설정하는 식입니다.
대한임상화학회 검사표준화및질향상연구위원회에서도 국내 실정에 맞는 임상화학검사 항목별 분석수행사양(APS) 목표치를 제시하고 있으며, 여기에는 각 검사 항목별 허용 바이어스 목표치가 포함되어 있습니다 [14, 15]. 이러한 국내 전문가 그룹의 권고안 역시 CD 설정에 중요한 참고 자료가 될 수 있습니다.
다음 표는 몇 가지 일반적인 임상화학 검사 항목에 대한 허용 바이어스 목표치의 예시를 보여줍니다 (값은 출처나 설정 기준에 따라 다를 수 있으므로 실제 적용 시에는 검사실의 정책과 최신 자료를 따라야 합니다). 이러한 값들이 CD 설정의 기반이 될 수 있습니다.
| 검사 항목 | 허용 바이어스 목표 예시 (생물학적 변동 - 바람직한 수준 기준) | 허용 바이어스 목표 예시 (CLIA 기준의 일부 활용 등) |
|---|---|---|
| 포도당 (Glucose) | ≤ 2.2% | ≤ 5% 또는 ≤ 3 mg/dL |
| 총 콜레스테롤 (Total Cholesterol) | ≤ 3.0% | ≤ 5% |
| HDL 콜레스테롤 | ≤ 4.0% | ≤ 15% |
| 중성지방 (Triglyceride) | ≤ 5.8% | ≤ 12.5% |
| 나트륨 (Sodium, Na) | ≤ 0.2% | ≤ 2 mmol/L |
| 칼륨 (Potassium, K) | ≤ 1.8% | ≤ 0.25 mmol/L |
| 염소 (Chloride, Cl) | ≤ 0.8% | ≤ 2.5% |
| 칼슘 (Calcium, Ca) | ≤ 0.8% | ≤ 0.5 mg/dL |
| 크레아티닌 (Creatinine) | ≤ 3.0% | ≤ 7.5% 또는 ≤ 0.15 mg/dL |
| 요소 (Urea/BUN) | ≤ 5.8% | ≤ 4.5% 또는 ≤ 1 mg/dL |
| 알부민 (Albumin) | ≤ 1.4% | ≤ 5% |
| 총 단백 (Total Protein) | ≤ 1.3% | ≤ 5% |
| AST (SGOT) | ≤ 5.8% | ≤ 10% |
| ALT (SGPT) | ≤ 7.8% | ≤ 10% |
| 빌리루빈 (Bilirubin) | ≤ 11.2% (Total) | ≤ 10% 또는 ≤ 0.2 mg/dL |
| 갑상선자극호르몬 (TSH) | ≤ 7.0% | (다양한 기준 존재) |
| HbA1c (당화혈색소) | ≤ 1.5% | ≤ 3% ~ 5% |
(주: 위 표의 값은 예시이며, 실제 적용 시에는 반드시 최신 EFLM 데이터베이스, CLIA 규정, 국내외 전문가 권고안 등을 종합적으로 검토하여 검사실 상황에 맞게 결정해야 합니다. 특히 생물학적 변동 기반 목표는 %값으로 제시되지만, 낮은 농도에서는 절대값 기준 적용이 필요할 수 있습니다.)
중요한 것은, 어떤 방법을 사용하든 CD 값은 합리적인 근거를 바탕으로 신중하게 결정되어야 하며, 왜 그 값을 CD로 설정했는지에 대한 명확한 이유와 기록이 있어야 한다는 점입니다. 또한, CD 값은 고정된 것이 아니라, 새로운 임상적 요구사항이나 검사 기술의 발전, TEa 목표치의 변경 등에 따라 주기적으로 검토되고 개정될 필요가 있습니다. 올바른 CD 설정이야말로 CLSI EP26 방법론을 효과적으로 활용하기 위한 핵심적인 전제 조건임을 반드시 명심해야 합니다.
CLSI EP26-Ed2 간소화 방법론
방법론의 핵심 원리
자, 이제 우리는 시약 로트 변경 검증의 중요성과 두 가지 핵심 개념, 즉 검사실 내 비정밀도(CVwL)와 임계 차이(CD)에 대해 이해했습니다. 그렇다면 CLSI EP26-Ed2 지침은 이 두 개념을 어떻게 활용하여 로트 변경 검증을 위한 최소 검체 수를 결정하는 간소화된 방법을 제공할까요? 그 핵심 원리는 생각보다 직관적입니다.
핵심 원리는 바로 신호 대 잡음비(Signal-to-Noise Ratio) 개념과 유사합니다. 여기서 신호(Signal)'는 우리가 감지하고자 하는 실제 로트 간 차이 중 임상적으로 유의미한 수준의 차이, 즉 임계 차이(CD)에 해당하고, 잡음(Noise)'은 검사 시스템 자체의 고유한 변동성, 즉 검사실 내 비정밀도(CVwL)에 해당합니다 [12, 15].
생각해보세요. 아주 조용한 방에서는 작은 속삭임(신호)도 쉽게 알아들을 수 있습니다. 하지만 시끄러운 경기장 한복판에서는 바로 옆 사람의 외침(신호)조차 듣기 어려울 수 있습니다. 마찬가지로, 검사 시스템의 비정밀도(잡음, CVwL)가 매우 낮다면(매우 정밀하다면), 로트 간의 작은 차이(신호, CD)라도 비교적 쉽게, 적은 수의 검체만으로도 통계적으로 유의미하게 감지해낼 수 있습니다. 왜냐하면 측정 결과의 '흔들림' 자체가 작기 때문에, 작은 차이라도 그 흔들림을 넘어 눈에 띄기 때문입니다 [12, 15].
반대로, 검사 시스템의 비정밀도(잡음, CVwL)가 크다면(정밀도가 낮다면), 로트 간의 상대적으로 큰 차이(신호, CD)가 발생해야만 그 차이가 단순한 무작위 변동인지 아니면 실제 로트 간의 차이인지 구별할 수 있습니다. 즉, 잡음이 크면 그 속에서 진짜 신호를 찾아내기 위해 더 많은 데이터(검체)를 측정해야 한다는 것입니다 [12, 15].
CLSI EP26-Ed2 지침은 바로 이 원리를 적용합니다. 검사실에서 설정한 임계 차이(CD)와 해당 검사 방법의 검사실 내 비정밀도(CVwL) 사이의 상대적인 크기(비율)를 계산하여, 통계적으로 의미 있는 로트 간 차이(CD 수준의 차이)를 일정한 확률(통계적 검정력, Power)로 탐지해내기 위해 필요한 최소 검체 수(N)를 결정하는 것입니다 [8, 17].
만약 CD가 CVwL에 비해 매우 크다면 (CD / CVwL 비율이 높다면), 이는 신호'가 잡음'보다 훨씬 크다는 의미입니다. 따라서 로트 간에 임상적으로 중요한 차이(CD)가 발생했을 경우, 이를 적은 수의 검체만으로도 높은 확률로 발견할 수 있습니다. 따라서 필요한 최소 검체 수(N)는 적어집니다 [12, 15, 8]. 예를 들어, CD가 10%이고 CVwL이 1%라면, 그 비율은 10입니다. 이 경우, 아주 적은 수의 샘플(예: 1개 또는 몇 개)만으로도 충분할 수 있습니다 [8].
만약 CD가 CVwL과 비슷하거나 더 작다면 (CD / CVwL 비율이 낮다면), 이는 신호'가 잡음'과 비슷하거나 오히려 작다는 의미입니다. 즉, 우리가 발견해야 할 로트 간 차이(CD)가 검사 자체의 변동성(CVwL) 속에 묻혀버릴 가능성이 높습니다. 따라서 이러한 작은 차이를 우연한 변동과 구별하고 확실하게 탐지해내기 위해서는 더 많은 검체(N)를 측정하여 통계적 검정력을 확보해야 합니다 [12, 15, 8]. 예를 들어, CD가 2%이고 CVwL도 2%라면 비율은 1입니다. 이 경우, 수십 개의 샘플이 필요할 수도 있습니다 [26].
또한, CLSI EP26-Ed2 지침은 검사실 내 비정밀도(CVwL) 대비 반복 정밀도(CVr)의 비율도 고려합니다. 구체적으로, CVwL에 비해 CD가 작거나, 또는 CVwL 대비 CVr이 작은 경우에는 필요한 검체 수가 많아진다고 언급합니다 [12, 15]. 검사실 내 비정밀도(CVwL) 대비 반복 정밀도(CVr)의 비율이 작다는 것은, 검사실 내 총 비정밀도(CVwL) 중에서 단기적인 반복 측정에 의한 변동성(CVr)이 차지하는 비중이 작다는 의미입니다.
이는 즉, 장기적인 요인(예: 다른 검사자, 다른 보정 주기 등)에 의한 변동성이 상대적으로 크다는 것을 시사합니다. 이러한 장기적 변동 요인이 클 경우, 로트 간의 미묘한 차이를 안정적으로 감지하기 어려울 수 있으므로 더 많은 검체가 필요할 수 있다는 논리로 해석될 수 있습니다. EP26 지침에서는 이 비율(Sr/Swrl 또는 CVr/CVwL)을 고려하여 검체 수 산정 및 판정 기준(rejection limit)을 조정하는 구체적인 표나 계산법을 제공합니다 [8, 17].
요약하자면, CLSI EP26-Ed2의 간소화된 방법은 다음의 핵심 원리에 기반합니다. 첫째, 검사실은 각 검사 항목에 대해 임상적으로 허용 가능한 로트 간 최대 차이(CD)를 정의합니다. 둘째, 검사실은 해당 검사 방법의 장기적인 변동성, 즉 검사실 내 비정밀도(CVwL)와 단기 변동성인 반복정밀도(CVr)를 파악합니다.
셋째, CD와 CVwL, 그리고 CVr/CVwL 비율을 비교하여, 설정된 CD 수준의 차이를 특정 확률(Power)로 탐지하는 데 필요한 최소 검체 수(N)를 결정하는 것입니다 [8, 17]. 마지막으로, 이 최소 검체 수(N)를 사용하여 실제 로트 변경 검증을 수행하고, 관찰된 차이가 미리 정해진 판정 기준(rejection limit, CD를 기반으로 계산됨)을 넘는지 평가하여 새 로트의 수용 여부를 결정합니다 [8, 13].
이러한 접근 방식은 각 검사의 특성(임상적 중요도와 분석적 성능)을 반영하여 필요한 검체 수를 합리적으로 결정함으로써, 불필요한 검사를 줄이고 자원을 효율적으로 사용하면서도 로트 간의 임상적으로 중요한 차이를 놓치지 않도록 설계되었습니다 [13, 16]. 이것이 바로 EP26 지침이 '간소화된' 방법이라고 불리는 이유입니다. 하지만 이 '간소화'는 검증 실행 단계에 해당하며, 이 방법을 적용하기 위한 초기 설정(Setup) 단계에서는 CD 설정, CVwL 및 CVr 파악 등 신중한 고려와 계산이 필요합니다 [13, 16, 27].
검증 절차 사전 준비
CLSI EP26-Ed2 지침의 간소화된 방법을 적용하여 로트 변경 검증에 필요한 최소 검체 수를 산정하고 평가를 수행하는 구체적인 단계별 과정은 크게 사전 준비 단계(Setup)와 실제 평가 단계(Evaluation)로 나눌 수 있습니다 [13, 16, 27]. 사전 준비 단계는 각 검사 항목에 대해 한 번만 수행하면 되며, 이 단계에서 결정된 사항들을 바탕으로 실제 로트 변경 시에는 간단하고 신속하게 평가를 진행할 수 있습니다 [13, 16, 27].
비정밀도 파악 및 CD 값 확인
가장 먼저 해야 할 일은 평가 대상 검사 방법의 '잡음' 수준, 즉 검사실 내 비정밀도(CVwL)와 반복 정밀도(CVr)를 정확하게 파악하는 것입니다. 이는 로트 간 차이를 평가하는 기준선이 되기 때문에 매우 중요합니다.
비정밀도 값은 다양한 방법으로 얻을 수 있습니다. 과거 검증 데이터 활용입니다. 검사실에서 해당 검사 방법을 처음 도입할 때 수행했던 정밀도 검증 실험 데이터(예: CLSI EP05 또는 EP15 지침에 따른 실험)를 활용할 수 있습니다 [6, 9, 11, 28]. CLSI EP15-A3 지침은 5일 동안 매일 5회 반복 측정하는 프로토콜(총 25회 측정)을 통해 CVr과 CVwL을 추정하는 방법을 제시하며, 많은 검사실에서 이 방법을 사용합니다 [9, 11, 19, 20, 31]. EP05는 더 장기간(최소 20일)에 걸쳐 더 많은 데이터를 요구하는 더 엄격한 방법입니다 [9, 28, 29, 31]. 이러한 실험 데이터를 통계적으로 분석(주로 분산분석, ANOVA 사용)하여 CVwL과 CVr 추정치를 계산합니다 [6, 9, 11, 28].
장기간의 내부정도관리(IQC) 데이터 활용입니다. 검사실에서 매일 또는 주기적으로 측정하는 내부정도관리 물질의 장기간(예: 최소 6개월 이상) 데이터를 이용하여 CVwL을 추정할 수도 있습니다 [5]. 이는 실제 검사실 운영 환경에서의 변동성을 잘 반영할 수 있다는 장점이 있습니다. CVr은 IQC 데이터를 활용하여 추정하기 어려울 수 있습니다.
또한 제조사 제공 정보 활용입니다. 검사 장비 또는 시약 제조사가 제공하는 제품 설명서나 성능 평가 자료에 CVwL과 CVr 값이 제시되어 있는 경우 이를 참고할 수 있습니다 [8, 19, 28]. 하지만 이는 제조사의 이상적인 조건에서 얻어진 값일 수 있으므로, 실제 검사실 환경에서의 성능과 차이가 있을 수 있다는 점에 반드시 유의해야 합니다. 가능하면 검사실 자체 데이터로 검증하는 것이 가장 좋습니다.
비정밀도는 검사 결과의 농도 수준에 따라 달라지는 경우가 많습니다 (예: 저농도에서는 CV가 크고, 고농도에서는 작은 경향). 따라서 로트 변경 검증은 임상적으로 중요한 결정을 내리는 농도 수준(Medical Decision Points, MDPs)에서 수행되어야 합니다 [7, 14, 17, 26]. 예를 들어, 당뇨 진단 기준 농도, 치료 약물 농도의 목표 범위 상한/하한 등이 MDP가 될 수 있습니다.
따라서 CVwL과 CVr 값도 각 MDP에 해당하는 농도에서의 값을 파악해야 합니다. 만약 해당 농도에서 직접 측정한 데이터가 없다면, 인접 농도의 데이터를 이용하거나 통계적 방법(예: 보간법)을 사용하여 추정할 수도 있습니다 [17]. 사용하는 CVwL 및 CVr 값은 현재 검사실의 실제 성능을 정확하게 반영해야 합니다. 오래된 데이터거나 검사 시스템에 변화(예: 장비 교체, 유지보수 상태 변경 등)가 있었다면 다시 평가해야 할 수 있습니다. 신뢰할 수 있는 비정밀도 데이터를 확보하는 것이 EP26 방법 적용의 가장 중요한 첫 단추입니다.
다음 단계는 새로운 로트와 기존 로트 간에 허용할 수 있는 최대 결과 차이, 즉 임계 차이(CD)를 설정하는 것입니다. 앞서 설명했듯이, 이는 통계적 기준이 아닌 임상적 유의성에 기반해야 합니다 [7, 13, 16]. CD 값은 각 검사 항목의 특성과 임상적 중요성을 고려하여 설정되어야 하며, 일반적으로 총 허용 오차(Total Allowable Error, TEa)의 허용 바이어스(Allowable Bias) 부분을 기반으로 설정됩니다 [12, 14, 15]. 검사실은 이러한 CD 설정에 대한 근거를 명확히 문서화하고, 각 MDP 농도에 해당하는 CD 값을 결정해야 합니다.
최소 검체 수 및 판정 기준 산정
이제 비정밀도 및 CD 값 확인 단계에서 얻은 CVwL 및 CVr 값과 정의한 CD 값을 이용하여, 로트 변경 검증에 필요한 최소 검체 수(N)와 새 로트 수용 여부를 판단할 기준(판정 기준(Rejection Limit)을) 결정합니다 [7, 8, 17, 18]. CLSI EP26-Ed2 지침은 이 과정을 돕기 위한 표(Table) 또는 계산 공식)을 제공합니다 [7, 8, 17, 18].


EP26 지침의 표나 공식은 기본적으로 두 가지 비율을 입력 값으로 사용합니다. 첫째는 CD / CVwL (또는 CD / SwL) 값입니다. 임계 차이를 검사실 내 비정밀도로 나눈 값이며, 이 값이 클수록 필요한 검체 수는 적어집니다. 둘째는 CVr / CVwL (또는 Sr / SwL) 값입니다. 반복 정밀도를 검사실 내 비정밀도로 나눈 값으로, 이 값은 판정 기준(Rejection Limit)을 조정하는 데 사용될 수 있습니다 [8, 17].
EP26 지침의 표(Table)에서 계산된 두 비율 값에 해당하는 행과 열을 찾아 교차하는 지점의 값을 읽으면, 특정 통계적 검정력(Statistical Power, 예: 90% 또는 95%)으로 CD 수준의 차이를 탐지하는 데 필요한 최소 검체 수(N)가 제시됩니다 [8, 17, 18]. 통계적 검정력이란, 실제로 로트 간 차이가 존재할 때 이를 '차이가 있다'고 올바르게 판단할 확률을 의미하며 [8, 18], 일반적으로 80%~90% 이상의 검정력을 목표로 합니다 [8, 18].
예를 들어, 특정 검사의 MDP에서 계산된 CD/CVwL 값이 6.0이고 CVr/CVwL 값이 0.6이라고 가정해 봅시다. EP26 지침의 해당 표(가상의 예시)를 찾아보면, 이 조건에서 95%의 검정력으로 로트 간 차이를 검출하기 위해 필요한 최소 검체 수(N)가 '1'개라고 나올 수 있습니다 [8]. 또 다른 검사에서는 동일한 비율 계산 결과, 검체 수(N)가 '23'개로 나올 수도 있습니다 [26].
EP26 지침은 단순히 '차이가 있다/없다'만 보는 것이 아니라, 관찰된 로트 간 평균 차이가 어느 정도일 때 새 로트를 거부(reject)할지에 대한 구체적인 기준, 즉 판정 기준(Rejection Limit)을 설정하도록 합니다. 이 판정 기준(Rejection Limit)은 일반적으로 CD 값에 특정 계수(factor)를 곱하여 계산됩니다. 이 계수는 앞서 계산한 CD/CVwL 비율과 CVr/CVwL 비율, 그리고 선택한 검체 수(N)에 따라 EP26 지침의 표나 공식에서 찾아 적용합니다 [8, 17].
예를 들어, 앞선 예시에서 검체 수(N)가 1개로 결정되었고, CD/CVwL=6.0, CVr/CVwL=0.6 조건에 해당하는 판정 기준 계수가 0.6이라면, 판정 기준(Rejection Limit)은 0.6 × CD가 됩니다 [8]. 즉, 1개의 검체를 측정했을 때 두 로트 간의 차이가 0.6 × CD를 초과하면 새 로트를 거부하고, 그 이하라면 수용하는 것입니다. 만약 검체 수(N)가 여러 개라면, N개 검체에서 얻은 평균 차이를 이 판정 기준과 비교하게 됩니다. CVr/CVwL 비율이 높을수록(즉, 단기 변동성이 클수록) 판정 기준이 약간 더 완화될 수 있습니다(즉, Factor 값이 커질 수 있음).
각 검사 항목 및 MDP별로 결정된 최소 검체 수(N)와 판정 기준(Rejection Limit)을 명확하게 문서화하여, 실제 로트 변경 시 쉽게 참조하고 적용할 수 있도록 준비합니다 [13, 21]. 체크리스트나 스프레드시트 형태가 유용할 수 있습니다 [13].
검증 절차 실제 평가
사전 준비 단계가 완료되었다면, 이제 새로운 시약 로트가 입고되었을 때 실제 평가 단계(Evaluation)를 수행하는 것은 매우 간단합니다 [13, 16, 27].
첫째, 3단계에서 결정된 최소 검체 수(N)만큼의 신선한 환자 검체를 준비합니다. 이 검체들은 평가하려는 MDP 농도 수준에 해당하는 검체들이어야 합니다. 이상적으로는 각기 다른 환자의 검체를 사용하는 것이 좋습니다.
둘째, 준비된 N개의 환자 검체를 기존(현재 사용 중인) 로트 시약과 새로운 로트 시약으로 각각 측정합니다. 셋째, 측정은 가능한 한 동일한 조건(예: 동일 장비, 동일 검사자, 짧은 시간 간격)에서 수행하는 것이 좋으며, 각 검체에 대해 새 로트 결과와 기존 로트 결과 간의 차이(Difference)를 계산하고 N개의 검체에 대한 평균 차이(Mean Difference)를 구합니다.
넷째, 계산된 평균 차이를 3단계에서 미리 설정해 둔 판정 기준(Rejection Limit)과 비교합니다. 만약 평균 차이 ≤ 판정 기준: 이라면 관찰된 로트 간 차이가 허용 범위 이내이므로, 새로운 시약 로트는 수용 가능(acceptable)합니다. 반대로 평균 차이 > 판정 기준: 이라면 관찰된 로트 간 차이가 허용 범위를 초과하므로, 새로운 시약 로트는 거부(rejected)해야 합니다. 이 경우, 문제의 원인을 조사하고(예: 시약 자체의 문제인지, 운송/보관 문제인지, 검사 시스템 문제인지 등), 필요한 경우 제조사에 연락하거나 추가적인 검증 절차를 진행해야 합니다.
이처럼 CLSI EP26-Ed2의 간소화된 접근법은, 초기 설정 단계에 노력을 집중함으로써 실제 평가 단계에 로트 변경 시에는 최소한의 검체와 간단한 계산만으로 신속하고 효율적인 평가를 가능하게 합니다.
아래 표는 EP26 절차를 요약한 것입니다.
| 단계 | 활동 내용 | 시점 | 주요 결과물/고려사항 |
|---|---|---|---|
| 1. 비정밀도 결정 | 검사실 내 비정밀도(CVwL) 및 반복 정밀도(CVr)를 각 MDP에서 파악 | 사전 준비 | 정확한 CVwL, CVr 값 (과거 검증 데이터, IQC 데이터, 제조사 정보) |
| 2. CD 정의 | 각 MDP에서 임상적으로 허용 가능한 최대 로트 간 차이(CD) 설정 (주로 TEa 기반 허용 바이어스 활용) | 사전 준비 | 각 MDP별 CD 값 (임상적 중요성 반영, 근거 기반 설정) |
| 3. N 및 판정 기준 결정 | CD/CVwL, CVr/CVwL 비율 및 목표 검정력을 이용하여 EP26 지침(표/공식)에 따라 최소 검체 수(N)와 판정 기준(Rejection Limit) 결정 | 사전 준비 | 각 MDP별 N, 판정 기준 (문서화 필수) |
| 4. 실제 평가 | 1) N개의 환자 검체 준비 (MDP 농도) 2) 기존 로트와 새 로트로 각각 측정 3) 평균 차이 계산 4) 판정 기준과 비교하여 수용/거부 결정 |
실제 로트 변경 시 | 새 로트 수용 또는 거부 결정 |
물론, 이 방법이 모든 상황에 완벽한 해결책은 아닙니다. 정확한 비정밀도 데이터 확보의 어려움, 적절한 CD 설정의 어려움, 때로는 여전히 많은 수의 검체가 요구될 수 있다는 점 등은 한계점으로 지적되기도 합니다 [26]. 하지만 기존의 주관적이거나 비표준화된 로트 검증 방식에 비해 통계적으로 훨씬 견고하고, 자원 효율적인 대안을 제공한다는 점에서 그 가치가 매우 높다고 할 수 있습니다. 이 '간소화'는 검증 실행 단계에 해당하며, 이 방법을 적용하기 위한 초기 설정(Setup) 단계에서는 CD 설정, CVwL 및 CVr 파악 등 신중한 고려와 계산이 필요합니다 [13, 16, 27].
EP26 방법론 적용 및 실제 사례
방법론의 장점과 단점
진단검사의학 분야에서 정확하고 신뢰할 수 있는 검사 결과를 제공하는 데 필수적인 시약 로트 변경 검증에 있어 CLSI EP26-Ed2 지침은 간소화된 로트 변경 검증 방법은 많은 검사실에 유용한 도구를 제공하지만, 동시에 몇 가지 고려해야 할 점과 잠재적인 단점도 존재합니다. 이 방법론을 실제 검사실에 성공적으로 적용하기 위해서는 이러한 장단점을 명확히 이해하고, 발생 가능한 문제점들에 대해 미리 대비하는 것이 중요합니다.
이 방법론의 가장 큰 장점은 효율성 및 자원 절약입니다. 통계적으로 산출된 최소한의 필요 검체 수를 사용한다는 점입니다 [13, 16]. 기존의 비표준화된 방식(예: 무조건 특정 개수(5개 또는 10개)의 검체 사용)에 비해, 검사 항목의 성능(비정밀도)과 임상적 요구사항(CD)을 고려하여 과학적으로 필요한 최소한의 검체만 사용하므로, 시간, 시약, 인력 등 검사실 자원을 절약할 수 있습니다 [13]. 특히 로트 변경이 빈번한 검사실에서는 이러한 효율성 증대가 큰 도움이 될 수 있습니다 [12, 15].
둘째, 통계적 견고성입니다. 이 방법은 단순한 경험이나 관행이 아닌, 통계적 원리(신호 대 잡음비, 통계적 검정력)에 기반하여 설계되었습니다 [8, 18, 27]. 즉, 설정된 임계 차이(CD) 수준의 로트 간 차이를 특정 확률(Power)로 탐지할 수 있도록 과학적으로 설계되었으므로, 주관적인 판단에 의존하는 것보다 객관적이고 신뢰할 수 있는 결과를 제공합니다.
셋째, 표준화된 절차를 제공합니다. CLSI EP26 지침은 로트 변경 검증을 위한 명확하고 표준화된 절차를 제공합니다 [16, 27]. 초기 설정(Setup) 단계와 실제 평가(Evaluation) 단계를 구분하고, 각 단계에서 수행해야 할 활동과 고려사항을 구체적으로 제시함으로써, 검사실 간 또는 검사자 간의 평가 절차 일관성을 높이는 데 기여합니다. 이는 검사실 품질 관리 수준을 향상시키는 데 중요한 요소입니다.
넷째, 임상적 유의성 고려입니다. 단순히 통계적 차이 유무만을 보는 것이 아니라, 임상적으로 허용 가능한 차이(CD)를 기준으로 설정하여 평가하므로, 환자 진료에 실질적으로 의미 있는 로트 간 차이를 관리하는 데 초점을 맞춥니다 [7, 13, 16]. 이는 불필요한 로트 거부를 줄이고, 정말로 문제가 될 수 있는 차이에 집중하게 해줍니다.
하지만 이 방법론에는 몇 가지 단점과 고려사항이 있습니다. 첫째, 초기 설정의 복잡성입니다. 실제 평가 단계는 간소화되었지만, 이를 위한 초기 설정(Setup) 단계는 상당한 노력과 전문 지식을 요구합니다 [13, 16, 27]. 정확한 검사실 내 비정밀도(CVwL) 및 반복 정밀도(CVr) 데이터 확보, 임상적 의미를 반영한 적절한 임계 차이(CD) 설정 등은 신중한 분석과 판단이 필요하며, 때로는 어려움을 겪을 수 있습니다 [26]. 특히 신뢰할 수 있는 비정밀도 데이터가 부족하거나, CD 설정에 대한 명확한 근거를 찾기 어려운 경우 적용에 한계가 있을 수 있습니다.
둘째, 필요 검체 수가 여전히 많을 수 있음입니다. '간소화된' 방법이라고는 하지만, 검사 방법의 비정밀도(CVwL)가 크거나 설정된 CD 값이 매우 작은 경우에는 여전히 많은 수의 검체(N)가 필요할 수 있습니다 [12, 15, 26]. 일부 연구에서는 특정 검사 항목에 대해 EP26 프로토콜이 기존 방식보다 더 많은 검체를 요구하는 경우도 보고되었습니다 [26]. 이는 특히 검체 확보가 어려운 검사 항목이나 자원이 제한적인 검사실 환경에서는 부담이 될 수 있습니다.
셋째, CD 및 비정밀도 값의 정확성에 대한 의존성입니다. 이 방법의 유효성은 입력되는 CD 값과 CVwL, CVr 값의 정확성에 크게 좌우됩니다 [7, 26]. 만약 부정확하거나 오래된 비정밀도 데이터를 사용하거나, 임상적 현실과 동떨어진 CD 값을 설정한다면, 잘못된 검체 수가 산출되거나 로트 평가 결과 자체가 왜곡될 수 있습니다. 따라서 이러한 입력 값들을 주기적으로 검토하고 업데이트하는 노력이 반드시 필요합니다.
넷째, 모든 유형의 로트 간 차이를 감지하지 못할 수 있음입니다. EP26은 주로 로트 간의 평균적인 차이(bias shift)를 감지하는 데 초점을 맞춥니다. 만약 새로운 로트가 평균값은 유사하지만 비정밀도(imprecision)가 크게 변하거나, 특정 농도 범위에서만 비선형적인(non-linear) 차이를 보이는 등 다른 유형의 성능 변화가 발생한다면, EP26 방법만으로는 이를 충분히 감지하지 못할 수도 있습니다. 따라서 EP26 검증 결과가 수용 가능하더라도, 지속적인 내부정도관리(IQC) 및 외부정도관리(EQA) 참여를 통해 시약 성능을 모니터링하는 것이 중요합니다.
다섯째, 통계적 가정에 기반합니다 [18]. EP26 방법론은 특정 통계적 가정(예: 데이터의 정규 분포 등)에 기반할 수 있습니다. 만약 실제 데이터가 이러한 가정을 크게 벗어난다면, 결과의 타당성이 영향을 받을 수 있습니다. 여섯째, 판정 기준(Rejection Limit) 설정의 어려움입니다. CD 값과 비정밀도 비율을 이용하여 판정 기준을 정하는 과정이 다소 복잡하게 느껴질 수 있으며, EP26 지침의 표나 공식을 정확하게 해석하고 적용하는 데 주의가 필요합니다 [26].
결론적으로, CLSI EP26-Ed2의 간소화된 로트 변경 검증 방법은 많은 검사실에 유용한 도구가 될 수 있지만, 만능 해결책은 아닙니다. 이 방법의 성공적인 도입과 활용을 위해서는 장점을 최대한 살리면서 단점을 보완하기 위한 노력이 필요합니다. 즉, 정확한 성능 데이터 확보, 신중한 CD 설정, 주기적인 검토 및 업데이트, 그리고 다른 품질 관리 활동(IQC, EQA 등)과의 병행이 필수적입니다. 검사실은 이러한 점들을 충분히 고려하여 EP26 방법론을 자체 품질 관리 시스템 내에 효과적으로 통합해야 할 것입니다.
실제 적용 시나리오
이제 이론적인 설명을 넘어, CLSI EP26-Ed2 방법이 실제 검사실 환경에서 어떻게 적용될 수 있는지 몇 가지 가상의 시나리오를 통해 구체적으로 살펴보겠습니다. 이를 통해 각 단계의 의미와 결과 해석 방식을 더욱 명확하게 이해할 수 있을 것입니다. (주의: 아래 시나리오의 수치와 결과는 설명을 위한 예시이며, 실제 EP26 지침의 정확한 표나 계산 결과를 반영하는 것은 아닙니다.)
정밀도가 매우 높은 검사 (예: 혈청 나트륨(Na))
- 검사 항목: 혈청 나트륨 (Sodium, Na)
- 임상적 중요 농도(MDP)입니다. 135 mmol/L (정상 하한 근처), 145 mmol/L (정상 상한 근처)
1단계 (비정밀도 결정)입니다. 장기간 IQC 데이터 및 EP15 검증 결과, 140 mmol/L 근처 농도에서 다음과 같은 비정밀도 값을 얻었다고 가정합니다. 검사실 내 비정밀도(CVwL)는 0.5% (SwL = 0.7 mmol/L)이며, 반복 정밀도 (CVr)는 0.3% (Sr = 0.42 mmol/L)입니다.
2단계 (CD 정의)입니다. 나트륨은 생리적 변동이 매우 작고 임상적 해석에 정밀도가 중요합니다. 생물학적 변동 기반 '바람직한 성능' 기준에 따라 허용 바이어스를 계산하니 약 0.2%로 매우 작게 나왔습니다. 검사실에서는 이를 반영하여 CD를 0.5% (0.7 mmol/L @ 140 mmol/L)로 설정하기로 결정했습니다 (매우 엄격한 기준).
3단계 (N 및 판정 기준 결정)입니다. CD / CVwL 비율은 0.5% / 0.5% = 1.0이며, CVr / CVwL 비율은 0.3% / 0.5% = 0.6입니다. EP26 지침 표(가상의 예시)에서 CD/CVwL=1.0, CVr/CVwL=0.6 조건, 90% 검정력을 만족하는 최소 검체 수(N) = 30개가 필요하다고 나왔습니다.
또한, 해당 조건에서의 판정 기준 계수(Factor)는 0.2라고 가정합니다. 따라서 판정 기준(Rejection Limit)은 0.2 × CD = 0.2 × 0.5% = 0.1% (또는 0.14 mmol/L)로 결정됩니다.
4단계 (실제 평가)입니다. 새로운 나트륨 시약 로트가 입고되었습니다. 135-145 mmol/L 범위의 환자 검체 최소 검체 수(N)만큼의 즉 30개를 준비합니다. 각 검체를 기존 로트와 새 로트로 측정하여 30개의 차이(Difference)를 얻습니다. 30개 차이값의 평균(Mean Difference)을 계산하니 +0.08% (+0.11 mmol/L)가 나왔습니다.
판정: 평균 차이의 절대값(0.08%)이 판정 기준(Rejection Limit)(0.1%)보다 작습니다. 따라서 새로운 시약 로트는 수용 가능(Acceptable)합니다.
비정밀도가 상대적으로 큰 검사 (예: 특정 종양 표지자)
- 검사 항목: 종양 표지자 XYZ
- 임상적 중요 농도(MDP)입니다. 50 U/mL (진단적 Cut-off 근처)
1단계 (비정밀도 결정)입니다. 과거 검증 데이터 분석 결과, 50 U/mL 근처 농도에서 비정밀도는 다음과 같습니다. 검사실 내 비정밀도(CVwL)는 8.0% (SwL = 4.0 U/mL)이며, 반복 정밀도 (CVr)는 5.0% (Sr = 2.5 U/mL)입니다.
2단계 (CD 정의)입니다. 이 종양 표지자는 생물학적 변동도 크고, 임상적으로는 큰 변화 추이를 보는 것이 더 중요하다고 판단하여, CLIA 기준 등을 참고하여 CD를 15% (7.5 U/mL @ 50 U/mL)로 설정했습니다 (상대적으로 덜 엄격한 기준).
3단계 (N 및 판정 기준 결정)입니다. CD / CVwL 비율은 15% / 8.0% = 1.875이며, CVr / CVwL 비율은 5.0% / 8.0% = 0.625입니다. EP26 지침 표(가상의 예시)에서 CD/CVwL=1.875, CVr/CVwL=0.625 조건, 90% 검정력을 만족하는 최소 검체 수(N) = 10개가 필요하다고 나왔습니다.
해당 조건에서의 판정 기준 계수(Factor)는 0.4라고 가정합니다. 따라서 판정 기준(Rejection Limit)은 0.4 × CD = 0.4 × 15% = 6.0% (또는 3.0 U/mL)로 결정됩니다.
4단계 (실제 평가)입니다. 새로운 XYZ 시약 로트가 입고되었습니다. 약 50 U/mL MDP 농도의 환자 검체 최소 검체 수(N)만큼의 즉 10개를 준비합니다. 각 검체를 기존 로트와 새 로트로 측정하여 10개의 차이(Difference)를 얻습니다. 10개 차이값의 평균(Mean Difference)을 계산하니 -7.2% (-3.6 U/mL)가 나왔습니다.
판정: 평균 차이의 절대값(7.2%)이 판정 기준(Rejection Limit)(6.0%)보다 큽니다. 따라서 새로운 로트는 거부(Rejected)해야 합니다. 원인 분석 및 추가 조치가 필요합니다.
CD 설정 값 변경의 영향 (시나리오 2에서 CD만 변경)
상황: 시나리오 2와 동일한 종양 표지자 XYZ 검사(CVwL=8.0%, CVr=5.0%)에 대해, 검사실에서 품질 목표를 더 강화하기로 결정하고 CD를 10% (5.0 U/mL @ 50 U/mL)로 더 엄격하게 재설정했습니다.
3단계 (N 및 판정 기준 재결정)입니다. CD / CVwL 비율은 10% / 8.0% = 1.25이며, CVr / CVwL 비율은 5.0% / 8.0% = 0.625로 동일합니다. EP26 지침 표(가상의 예시)에서 CD/CVwL=1.25, CVr/CVwL=0.625 조건, 90% 검정력을 만족하는 최소 검체 수(N) = 25개로 증가했습니다 (CD가 작아지니 필요한 검체 수가 늘어남).
해당 조건에서의 판정 기준 계수(Factor)는 0.25라고 가정합니다. 따라서 판정 기준(Rejection Limit)은 0.25 × CD = 0.25 × 10% = 2.5% (또는 1.25 U/mL)로 더 엄격해졌습니다.
영향 분석: 이처럼 CD를 더 작게(엄격하게) 설정하면, 로트 간의 더 작은 차이도 유의미하게 감지해야 하므로 필요한 최소 검체 수(N)가 증가하고, 판정 기준(Rejection Limit)도 더 엄격해집니다. 이는 로트 변경 검증의 민감도를 높이지만, 검사에 필요한 자원 부담은 커지게 됩니다. 반대로 CD를 더 크게 설정하면 필요한 검체 수는 줄어들지만, 임상적으로 의미 있는 차이를 놓칠 위험은 증가합니다. 따라서 검사실은 이러한 민감도와 자원 효율성 사이의 균형을 신중하게 고려하여 CD 값을 설정해야 합니다.
이러한 시나리오들을 통해 볼 수 있듯이, CLSI EP26-Ed2 방법은 각 검사의 분석적 성능(CVwL, CVr)과 임상적 요구사항(CD)을 종합적으로 고려하여 과학적이고 합리적인 로트 변경 검증 절차를 제공합니다. 검사실은 이 지침을 올바르게 이해하고 적용함으로써, 제한된 자원 내에서 효과적으로 검사 품질을 관리하고 환자 안전을 확보하는 데 큰 도움을 받을 수 있을 것입니다.
결론
지금까지 우리는 진단검사의학 검사실에서 새로운 시약 로트를 도입할 때 왜 검증이 필요한지 그 중요성부터 시작하여, CLSI EP26-Ed2 지침이 제시하는 간소화된 로트 변경 검증 방법의 핵심 원리와 구체적인 적용 단계, 그리고 관련 개념들(비정밀도, 임계 차이, 총 허용 오차, 허용 바이어스 등)에 대해 깊이 있게 살펴보았습니다.
핵심 내용을 다시 한번 요약 정리하면 다음과 같습니다. 첫째, 로트 변경 검증의 필요성입니다. 시약은 로트별로 미세한 성능 차이가 있을 수 있으며, 이는 환자 검사 결과의 비일관성을 초래하여 잘못된 임상적 판단으로 이어질 수 있습니다. 따라서 새 로트 사용 전, 기존 로트와의 차이가 임상적으로 허용 가능한 범위 내에 있는지 반드시 검증해야 합니다 [7, 10, 13].
둘째, 핵심 개념 - 비정밀도(Imprecision)입니다. 모든 검사에는 무작위 오차로 인한 결과의 변동성, 즉 비정밀도가 존재합니다. 특히 장기간의 검사실 운영 환경에서의 총 비정밀도를 나타내는 검사실 내 비정밀도(CVwL)가 중요합니다 [6, 9, 12, 15]. 이는 로트 간 차이를 평가할 때의 '잡음(noise)' 수준에 해당합니다. 셋째, 핵심 개념 - 임계 차이(CD)입니다. 이는 통계적 유의성이 아닌 임상적 유의성에 기반하여, 허용 가능한 로트 간 최대 결과 차이를 검사실이 미리 설정하는 기준입니다 [7, 13, 15]. 일반적으로 총 허용 오차(Total Allowable Error, TEa)의 허용 바이어스(Allowable Bias) 부분을 기반으로 설정됩니다 [12, 14, 15]. 이는 감지해야 할 신호(signal)'의 크기에 해당합니다.
넷째, EP26 간소화 방법의 원리: CD(신호)와 CVwL(잡음)의 상대적인 크기(비율)를 비교하여, CD 수준의 로트 간 차이를 일정한 통계적 검정력(Power)으로 탐지하는 데 필요한 최소 검체 수(N)와 판정 기준(Rejection Limit)을 결정합니다 [8, 12, 15, 17]. CD/CVwL 비율이 클수록 N은 작아지고, 작을수록 N은 커집니다. 다섯째, 적용 단계입니다. 사전 준비 단계에서 각 검사 항목 및 MDP별로 CVwL, CVr, CD 값을 결정하고, 이를 바탕으로 EP26 지침(표/공식)을 이용해 최소 검체 수(N)와 판정 기준(Rejection Limit)을 미리 설정 및 문서화합니다 [13, 16, 27]. 실제 평가 단계에서는 이 최소 검체 수(N)를 이용해 새 로트와 기존 로트를 비교 측정하고, 평균 차이가 판정 기준(Rejection Limit)을 넘지 않으면 새 로트를 수용합니다 [13, 16, 27]. 여섯째, 장점과 한계입니다. EP26 방법은 효율성, 통계적 견고성, 표준화, 임상적 유의성 고려 등의 장점이 있지만, 초기 설정의 복잡성, 여전히 많은 검체 수가 필요할 수 있는 가능성, 입력 값 정확성에 대한 의존성 등의 한계점과 고려사항도 가지고 있습니다 [26].
결론적으로, CLSI EP26-Ed2 지침은 시약 로트 변경 검증이라는 필수적인 품질 관리 활동을 보다 과학적이고 효율적으로 수행할 수 있도록 돕는 매우 유용한 도구입니다. 하지만 이 지침을 성공적으로 활용하기 위해서는 다음 사항들을 반드시 명심하고 실천해야 합니다.
첫째, 정확한 데이터 확보입니다. 신뢰할 수 있는 최신 검사실 내 비정밀도(CVwL) 및 반복 정밀도(CVr) 데이터를 확보하는 것이 무엇보다 중요합니다. 필요하다면 정기적인 재평가를 통해 데이터를 업데이트해야 합니다.
둘째, 신중한 CD 설정입니다. 각 검사 항목의 임상적 중요성과 특성을 충분히 고려하고, 명확한 근거(TEa, 생물학적 변동, 전문가 의견 등)를 바탕으로 임계 차이(CD)를 신중하게 설정해야 합니다. 설정된 CD 값은 주기적으로 검토하고 필요시 개정해야 합니다.
셋째, 지침의 정확한 이해와 적용입니다. EP26 지침의 원리와 절차를 정확하게 이해하고, 제공되는 표나 공식을 올바르게 해석하여 적용해야 합니다.
넷째, 다른 품질 관리 활동과의 연계입니다. EP26 로트 변경 검증은 전체적인 검사실 품질 관리 시스템의 일부일 뿐입니다. 이 검증을 통과했더라도 일상적인 내부정도관리(IQC) 및 외부정도관리(EQA) 참여를 통해 지속적으로 검사 성능을 모니터링하고 예상치 못한 문제를 조기에 발견하려는 노력이 병행되어야 합니다.
마지막으로, 지속적인 교육과 전문가 협의입니다. 검사실 직원들이 EP26 지침과 관련 통계 개념을 충분히 이해할 수 있도록 지속적인 교육이 필요하며, CD 설정이나 결과 해석에 어려움이 있을 경우 진단검사의학과 전문의 등 전문가와 적극적으로 협의하는 자세가 중요합니다.
궁극적으로 시약 로트 변경 검증의 목표는 검사 결과의 일관성과 신뢰성을 확보하여 환자에게 최상의 진료를 제공하는 것에 있습니다 [2, 10]. CLSI EP26-Ed2 지침을 올바르게 이해하고 충실히 이행하는 것은 이러한 목표 달성에 크게 기여할 것이며, 검사실 품질 관리 수준을 한 단계 높이는 중요한 발걸음이 될 것입니다.
2025.04.21 - [임상미생물] - 호흡기 감염의 원인균과 검사실 진단방법
호흡기 감염의 원인균과 검사실 진단방법
여러분, 혹시 숨쉬는 것이 당연하다고 생각해 본 적 있으신가요? 맑은 공기를 들이마시고 내쉬는 이 단순한 행위가 우리 생명의 근원이라는 사실, 잠시 잊고 살 때가 많지요. 하지만 어느 날 갑
labdoctor.tistory.com
2025.04.20 - [임상미생물] - 혈액 배양시 오염균 판정 기준
혈액 배양시 오염균 판정 기준
여러분, 혹시 '패혈증'이라는 말을 들어보신 적 있으신가요? 뉴스에서 종종 접하거나, 주변에서 안타까운 이야기를 통해 들어보셨을 수도 있겠습니다. 패혈증은 세균이나 곰팡이 같은 미생물이
labdoctor.tistory.com
2025.04.18 - [진단면역] - 면역 검사에서의 항체 간섭 현상의 원인과 기전, 해결방법
면역 검사에서의 항체 간섭 현상의 원인과 기전, 해결방법
여러분은 혹시 건강검진이나 병원 진료 과정에서 혈액 검사나 소변 검사를 받아보신 경험이 있으실 겁니다. 이러한 검사들 중 상당수는 우리 몸의 면역 시스템, 특히 항원-항체 반응이라는 놀라
labdoctor.tistory.com
2025.04.18 - [분자진단] - 세균 동정을 위한 16s rRNA 염기서열방법의 목적, 원리, 적용
세균 동정을 위한 16s rRNA 염기서열방법의 목적, 원리, 적용
우리 주변 세상은 눈에 보이지 않는 작은 생명체, 바로 미생물로 가득 차 있습니다. 흙 한 줌에는 수십억 마리의 세균이 살고 있고, 우리 몸속, 특히 장에는 우리 몸 세포 수보다 훨씬 많은 미생
labdoctor.tistory.com
참고문헌
- Westgard JO. Basic QC Practices. 3rd ed. Madison, WI: Westgard QC, Inc.; 2010.
- Fraser CG. Biological Variation: From Principles to Practice. Washington, DC: AACC Press; 2001.
- Clinical and Laboratory Standards Institute (CLSI). Evaluation of Precision of Quantitative Measurement Procedures; Approved Guideline—Third Edition. CLSI document EP05-A3. Wayne, PA: CLSI; 2014.
- Clinical and Laboratory Standards Institute (CLSI). User Verification of Precision and Estimation of Bias; Approved Guideline—Third Edition. CLSI document EP15-A3. Wayne, PA: CLSI; 2014.
- Clinical and Laboratory Standards Institute (CLSI). User Evaluation of Acceptability of a Reagent Lot Change; Approved Guideline—Second Edition. CLSI document EP26-Ed2. Wayne, PA: CLSI; 2022. [12, 15, 16, 25, 27]
- Clinical and Laboratory Standards Institute (CLSI). Measurement Procedure Comparison and Bias Estimation Using Patient Samples; Approved Guideline—Third Edition. CLSI document EP09c. Wayne, PA: CLSI; 2018.
- Kallner A, et al. Strategies to set global analytical quality specifications in laboratory medicine. Scand J Clin Lab Invest. 1999;59(7):475-585. [13]
- Ricós C, et al. Current databases on biological variation: pros, cons and progress. Scand J Clin Lab Invest. 1999;59(7):491-500.
- The European Federation of Clinical Chemistry and Laboratory Medicine (EFLM). Biological Variation Database. [Accessed April 25, 2025]. Available from: https://biologicalvariation.eu/
- 김솔잎, 임지숙, 민원기. 임상화학검사 품질관리를 위한 분석수행사양 활용. Laboratory Medicine Online. 2023;13(4):255-266. [14, 15]
- Miller WG, et al. Bias in laboratory medicine: The dark side of the moon. Clin Chim Acta. 2023;547:117447. [1, 2]
- Friedecky B, et al. Guidelines for Allowable Total Error for Hematology Testing. American Society for Veterinary Clinical Pathology (ASVCP). 2017. [3]
- Shaw C, et al. Total Allowable Error (TEa): How Much Error Can Your Laboratory Allow?. AACC Clinical Chemistry Trainee Council. December 1, 2021. [4]
- Ganji SB, Revupalli S. Evaluation of Quality Assurance in a New Clinical Chemistry Laboratory by Six Sigma Metrics. J Clin Diagn Res. 2019;13(2):BC01-BC04. [5]
- Chen W. Intermediate Precision Evaluation. KeepNotes blog. May 19, 2021. [6]
- George R, et al. Comparison of Sample Size and Rejection Limits for Lot-to-Lot Verification Using Two Different Protocols. Natl J Lab Medicine. 2024;13(3):LC01-LC04. [7]
- Karahan O, Kılıcgun H. Sample size, power and effect size revisited: simplified and practical approaches in pre-clinical, clinical and laboratory studies. Biochem Med (Zagreb). 2020;30(2):020501. [8, 18]
- Westgard JO. CLSI EP15-A3: verification of precision and estimation of bias. Westgard QC website. [Accessed April 25, 2025]. [9]
- Li S, et al. Research and discussion on the evaluation scheme of reagent lot-to-lot differences in 16 chemiluminescence analytes, established by the EP26-A guidelines of the CLSI. J Clin Lab Anal. 2021;35(2):e23642. [17]
- 박민정. 정밀도 정확도 검정 중심으로. [프레젠테이션]. 2018. [28]
'정도관리 통계' 카테고리의 다른 글
| 내부정도관리에서 pooled CV의 계산과 적용방법 (0) | 2025.04.12 |
|---|---|
| 정도관리(QC) 오류 감소를 위한 시약 제조 및 관리 방법 (0) | 2025.04.06 |
| 환자 검체를 이용한 장비간 비교 검사 (CSLI EP09) 상세 분석 (0) | 2025.04.06 |
| Calibrator lot 변경, 시약 lot 변경, 정도관리 lot 변경이 QC chart의 shift를 유발하는 이유 (1) | 2025.04.06 |
| Calibrator lot 변경, 시약 lot 변경, QC lot 변경시 lot to lot (parallel test) 시행해야 하는가? (0) | 2025.04.06 |




댓글